For the 20/5/20 non-covalent submission see reply below
Fragmenstein places a followup compound to the hits as faithfully as possible regardless of horrid bond lengths and torsions and energy minimises in place.
As a consequence, it is not really a docking algorithm as it does not find the pose with the lowest energy
within a given volume. Consequently, it is a method to find how faithful is a given followup to the hits provided.
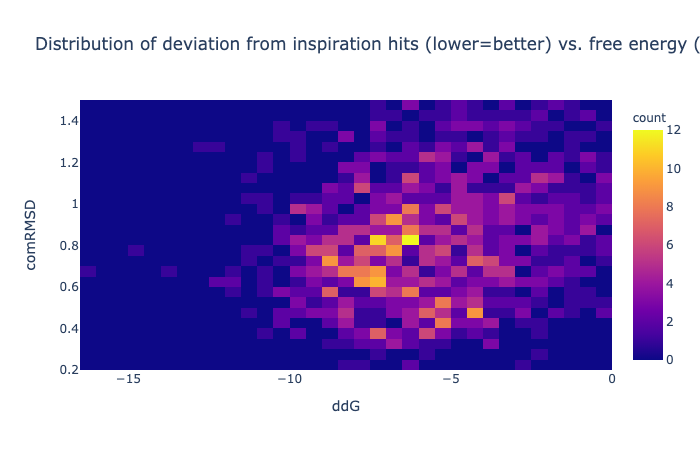
Hence the minimised pose should be assessed by the RMSD metric and the ∆∆G score used solely as a cutoff —lower than zero.
First the inspiration hits are merged regardless of horrid torsions and bond lengths.
Then the followup compound is mapped on (with appropriate atom changes) and novel parts added
Then the compound is minimised in the protein with constraints to the mapped atoms.
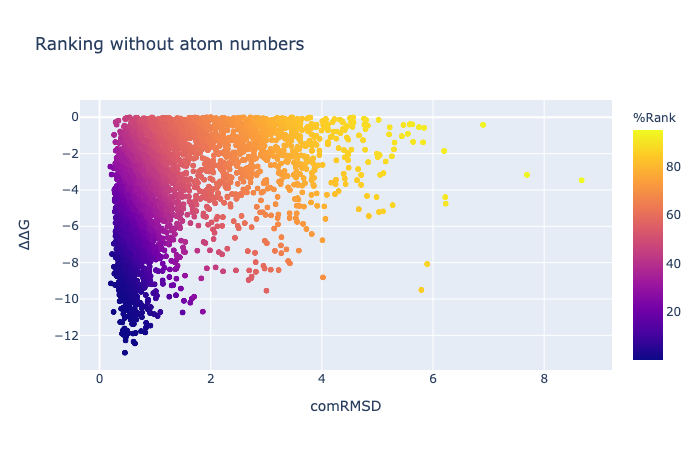
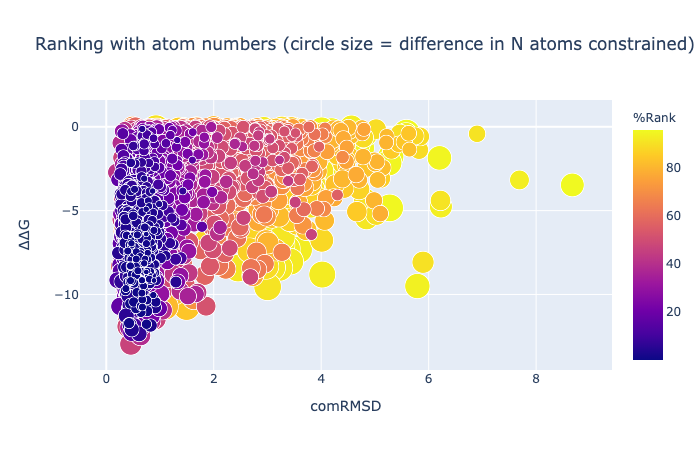
The combined RMSD is described here. The binding energy is the difference between the free energy of the ligand alone minus the free energy of a minimised unbound ligand (which is not necessarily zero). Units are kcal/mol. [Here are some examples of the meaning of kcal/mol](https://venus.sgc.ox.ac.uk/docs/venus_energetics. 1 kcal/mol is the same as 4.2 kJ/mol and in US units is 2.57 ft x lb /lbmol.
The data is incomplete and may submissions are missing, not due to error though (will be resolved shortly).
In the SMILES, the * (dummy/R) is the attachment point if covalent.
The warheads combinations are as follows:
CID as is = No change (formerly this would have the suffix _NIT1 _ACR1). If covalent the SMILES will have been changed to covalent.
_NIT to nitrile, _ACR to acrylamide, _CLR to chloracetimide, _VIN to vinylsulfoxide. These would have had the suffix followed by 2. Epoxyketones mentioned in a different post were not done as there was no discussion of these. For conversions see GitHub.
The RMSD can be calculated to any pose, even those generated by other methods…
Caveats
A lot of submission are designated as being “inspired” by x0072, whereas in reality they are based on fragment independent screen and chose the first compound because “None” was not an option. As some x0072 are legitimate and some other single compound inspirations may be bogus these should be taken with maximum suspicion.
The minimisation was done with Rosetta, but the protein or the neighbourhood of the ligand was neither minimised (relax) or repacked, in order to be maximally faithful to the submission hit.
Time permitting, I would like to use a merged density map to guide the the minimisation as a compromise.
Rosetta is tailored for protein and the atom types in its forcefields are more limited than would be desired, so certain groups may not be modelled well.
Fragmenstein assesses the fidelity to the hits.
However, there are three problems with the starting data.
Users select a large amount of inspiration hits, including ones that are relevant for other submissions in the set (as the inspiration hits is a common field).
Users select the wrong code
Users are not submitting compounds based on the hits (None is an option so x0072 is selected).
Fragmenstein has two steps, as mentioned, the first creating the reference positions.
Fragmenstein has now three options for this step:
no merging: The fragments are not merged, but the MCS mapping to a subset of the inspiration hits is maximised without violating positional overlap (unique one to one mapping within 2 Å), any hits that cause issues are excluded — very slow for 5+ hits ways
full merging: the hits are full merged based on positional overlap (used in previous dataset) —ring merger and too many compounds cause incomplete mapping
partial merging: the hits are partially merged and the best mapping is chosen.
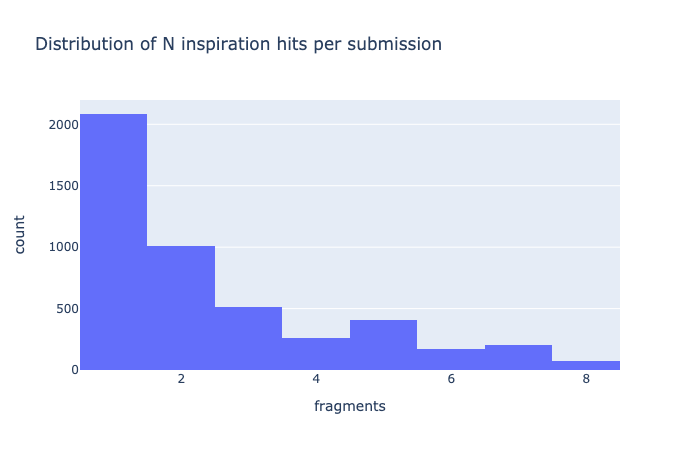
The later was introduced to better score cases where 4+ inspirations were given, which is indeed the group of compounds that this program was written for. Indeed, even if 44% of the submission have a single inspiration (1/8th of the submissions are based off x0072), 35% have 3+ or more inspirations.
In the previous set the protein was rigid, during energy minimisation of the merged fragment —the step where odd topological strains are ironed out. Herein, sidechains in the pocket were energy minimised before and after fragment energy minimisation.
To not cause local minima to cause trouble, the protein was energy minimised but constrained to electron density map (pyrosetta protocol). I used the PDB:6LU7 structure, as opposed to that of the largest hit because:
I am told the density maps generate by XChem differ from regular maps (cf. diamond site) —the CCP4 files from PDBe or gemmi coverted MTZ files seem to work fine though
And also @dmoustakas suggested to the modelling group that the structure in a pose around the native-substrate–like peptide is a better choice for docking.
Weighing the score
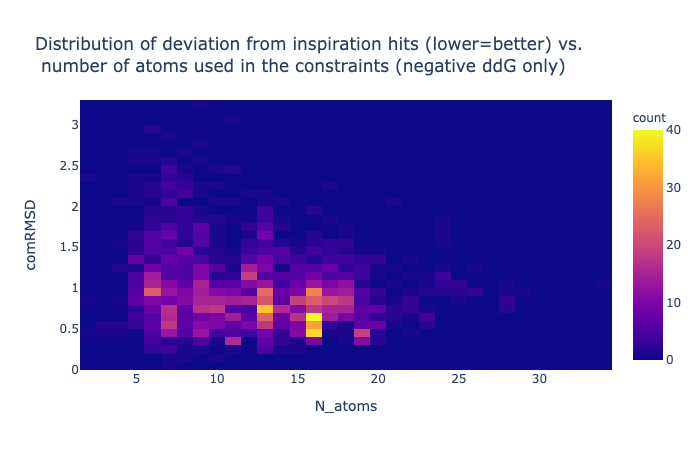
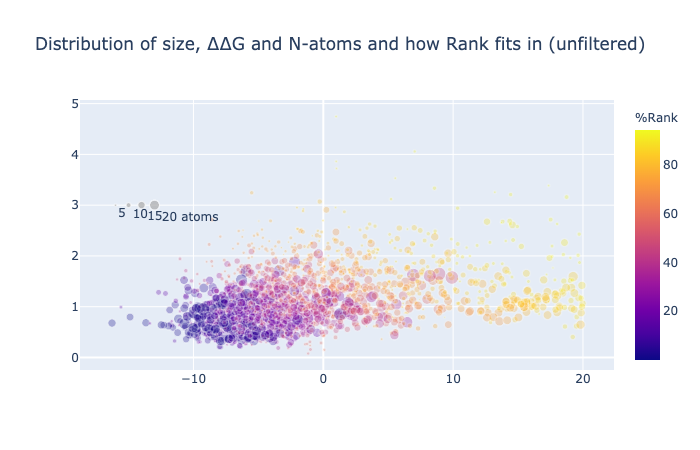
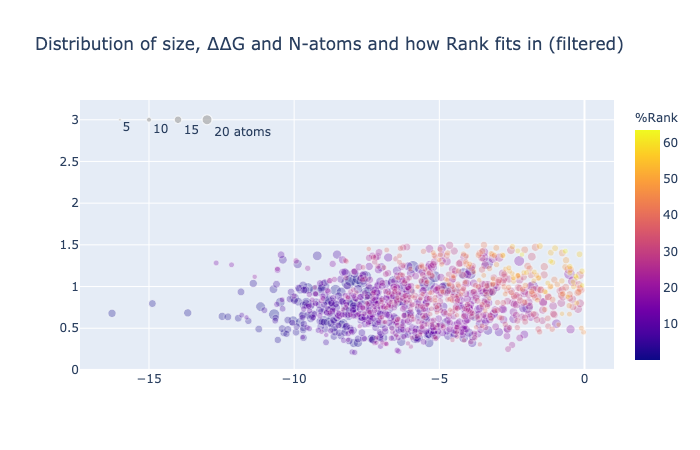
1/4 of the compounds have a negative binding energy (relative to unbound so negative is good), an RMSD against the concatenation of the inspiration hits less than 1.5 Å and have more than 10 atoms whose position was mapped.
This however, does not mean that the compounds that failed do not bind, it means that the compounds that failed are unlikely to bind in the way that was suggested by the inspiration hits.
The cutoff of 10 atoms minimum mapped stems from visual inspection of its distribution, basically the dataset contains a few compounds that are not actually inspired by the suggested fragments, so to remove the noise cause by cases where there is simply mapping say from a ring to a ring the spot where low cluster finishes was eyeballed to be ten. Although it should be said that the low number of mapped atom compounds score worse on average despite having less constraints.
The weights were empirically chosen to balance the distribution resulting in RMSD - ddG/5 - N_atoms/5.
Note that this is conjuction with the filtering mentioned.
This is about placing and scoring the fidelity of the followup submissions to the hits. Not generating submissions, for that see Fragmenstein: merging
Last week, @frankvondelft asked me to make it explicit which fragments were excluded by Fragmenstein.
Consequently, I re-run Fragmenstein with the newest dataset. I run it in
However, one major issue I have been having is merging the various values into a single rank, so input on how to better balance these is very welcome. I have the following values:
The minimisation is constrained by a given number of atoms, while others are not.
The ∆∆G is calculated using that score and is mainly to assess how happy the ligand is (greater than zero would indicate it’d rather be in solution).
This looks super cool… I’ll think a bit about the weighting - my inclination is to go with RMSD and filter by the other two, given the well-acknowledged inaccuracy of free energy estimation, use a cutoff to exclude the dross.
There may be bugs, see this one: HAN-NEW-5f56c3bc-3
I can’t find my six FRA-DIA compounds - is it personal?
Oh, also report number of merged fragments (not just their identity): that’d be my first sort criterion.
When I sort (in Michelanglo) by “comRMSD”, then the top-ranked are reams and reams of single-fragment designs - unsurprisingly.
(Once Fragalysis has the feature done (super-imminent!), this will be easy to filter; the simple sorting in Michelanglo (and most tables widgets) makes it harder to find.)
Sorry, what is wrong with it? I would have said it correctly excluded (in dark shades) the faux-inspirations.
This is an overly creative design, where atoms are added in between, so I am not surprised it has weird geometry. Interestingly, the warhead changes have different solutions —I did check how does the minimisation go with repeats and it was very close, but I did not try with something tricky like this, so it could be that different runs produce different results for nasty ones.
It is one with a mis-typed hit (x0425), which has not affected it—there are some 10 cases where the followup gets led astray by mis-typings.
However, this is a sorting problem, this one should not have floated to the top.
I think that ranking/sorting is the key problem.
FRA-DIA
Personal, no. Am I enjoying the irony… maybe.
FRA-DIA-8640f307 series failed, but that was expected as the indole was incorrect in the SMILES and I am fetching the csv from Github without fixing this. I discussed this & similar (Indole ring in SMILES) with Matt Robinson and the issue affects a small number of submissions and you did said to leave behind the entries where folk make mistakes…
—do you want me to resubmit FRA-DIA-8640f307 via PostEra with the correct bonding?
The other set were not in the data yet —I will do the new release now.
Better ranking
I will also score the followups that are crystallised and get the numbers as if they were placed and minimised, so the weights can be done by ML as @aaron.morris suggested.
Number of merged fragments will be a row/criterion/parameter/feature, thanks.
@mc-robinson, what must I do to get my FRA-DIA SMILES fixed? Can’t you just fix them in your database? Making me resubmit seems a bit extreme.
@matteoferla, you make a fair point, that we should review what counts as a “mistake”: I’ll comment in that thread, but that was a mistake of convention, not scientific understanding or intent.
(Do shift+refresh in chrome to ensure that the data is fresh).
As they are a mega-merger… They come first. Feels like a Kobayashi Maru scenario from Star Trek: if you cannot win cheat by altering the rules.
Odd. It is there in https://michelanglo.sgc.ox.ac.uk/data/13523b58-d0b1-4d05-9158-a8fd2be8465c (the latest: there are multiple instances of Fragmenstein results on Michelanglo) and was there before (is this a new/old CID issue?). However, it did not use x0387 as the submitter intended —I can see by eye that the atom rings don’t match up at all and one bond would have been greater than 3 Å, so it violated those two conditions.
No, the hits in light tints were used, while the hits in dark shades were excluded. The rank is low because of the non-mapped atoms, i.e. penalising creative submissions. It would never have matched. x0387 is a thiopene - C - piperidine, while the followup has two carbons before the piperidine and comes of the nitrogen off x0434 (everyone’s favourite urea bi-cycle), so it would have mapped solely if the thiopene weren’t there in x0387 and even then it would have underperformed as the linker would have done a 90ª angle.
The JOR-UNI-2fc98d0b-12 does not appear to be released yet??
Just a wee update. There have been several new submissions, many with states inspirations, as a consequence, I placed those with inspiration fragments with Fragmenstein.
I have not generated better weights by ML, unfortunately, for the ranking. I have continued to assign them based on what I perceive as important. ∆∆G favours large molecules —as is clear from the overly big Fragmenstein automerger results (https://michelanglo.sgc.ox.ac.uk/data/ba029bd3-28e3-41db-94e5-1b7c0dde3c3c). Hence the switch to ligand efficiency. However, this size agnostic metric needs some size bonus, so I added a constrained atom bonus that is rectangular hyperbola (“Michealis-Menten curve”) inflecting at 20 atoms, also novel atoms are not sought after here, so a penalty for too many novel atoms was also added. The ∆∆G score is relative to unbound in implicit waters, so greasy compounds with a high partition coefficient will score well, even if their true unbound ∆G is a membrane bound ∆G, not a water one.