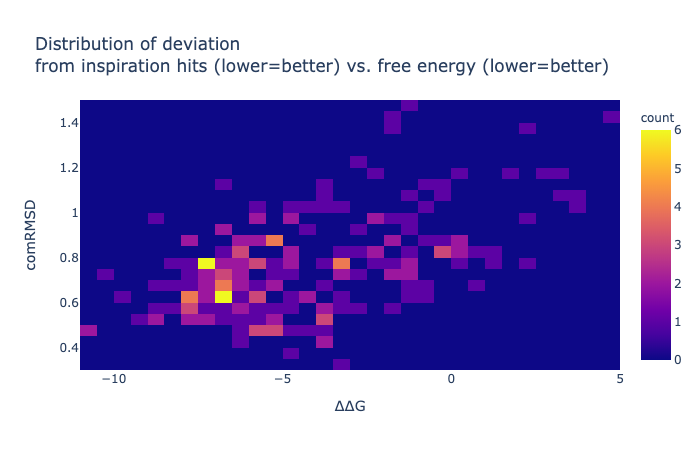
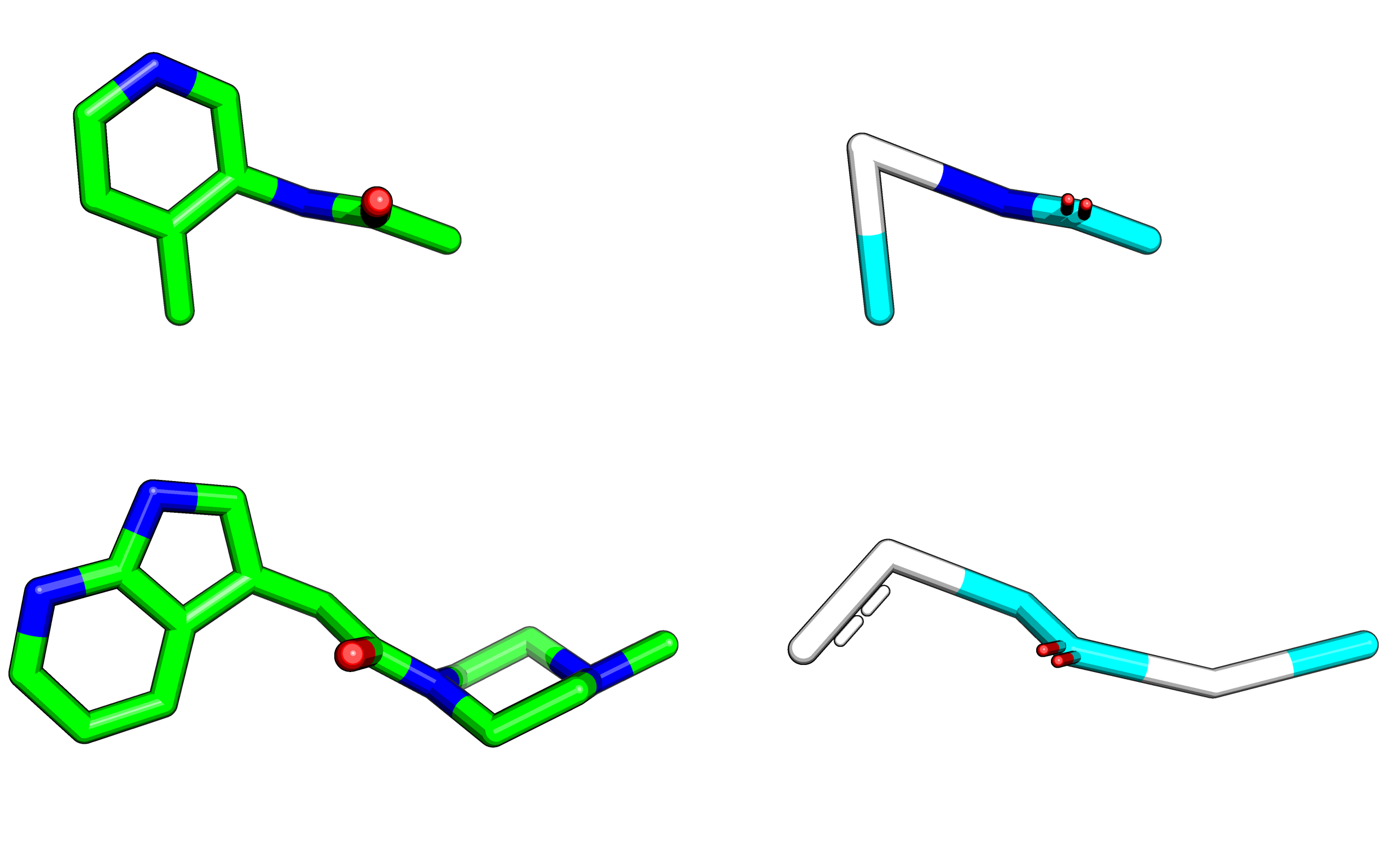
One way Fragmenstein maps submitted compounds to the inspirations is by merging them. Therefore with a few tweaks Fragmenstein merges hits happily*. Code: combine.pdf (159.4 KB)
Pairwise
Here is a table of pairwise combinations it predicted as proof of principle. The ranking was row.comRMSD + row['∆∆G']/10 - row.N_constrained_atoms /20 as it was only pairwise and as all inspirations are self-inspired (so way easier for it), the RMSD should get more weight.
I’ll do a more complex system later on. With more hits, the covalents and with filtering for only enumerated molecules in ZINC or similar.
*) Horrors lurk at the bottom of the table
These are solely based on one-to-one positional overlap within 2Å, therefore several compounds are impossible with a few Texas carbons —the horror show at the end of the table is actually entertaining to look at. Changing the formal charges and changing rings to non-aromatic were done when sanitization failed, but obviously this is not a real sanity check.
Rings
One thing that the Fragmenstein full-merger underperforms in is rings, this is especially true when there are 3+ rings, and differently sized, as suggested inspiration hits. As a consequently, I have been trialling out collapsing rings before the positional mapping (not used here), but the code struggles with a few corner cases.
I am hoping that this will make less horrors and more sensible molecules.
Cool! Do you have the poses in a Michelanglo view?
A few horrors lurk right at the top too, I’m afraid… 559, 861… but it’s a very cool start. Keep us posted!
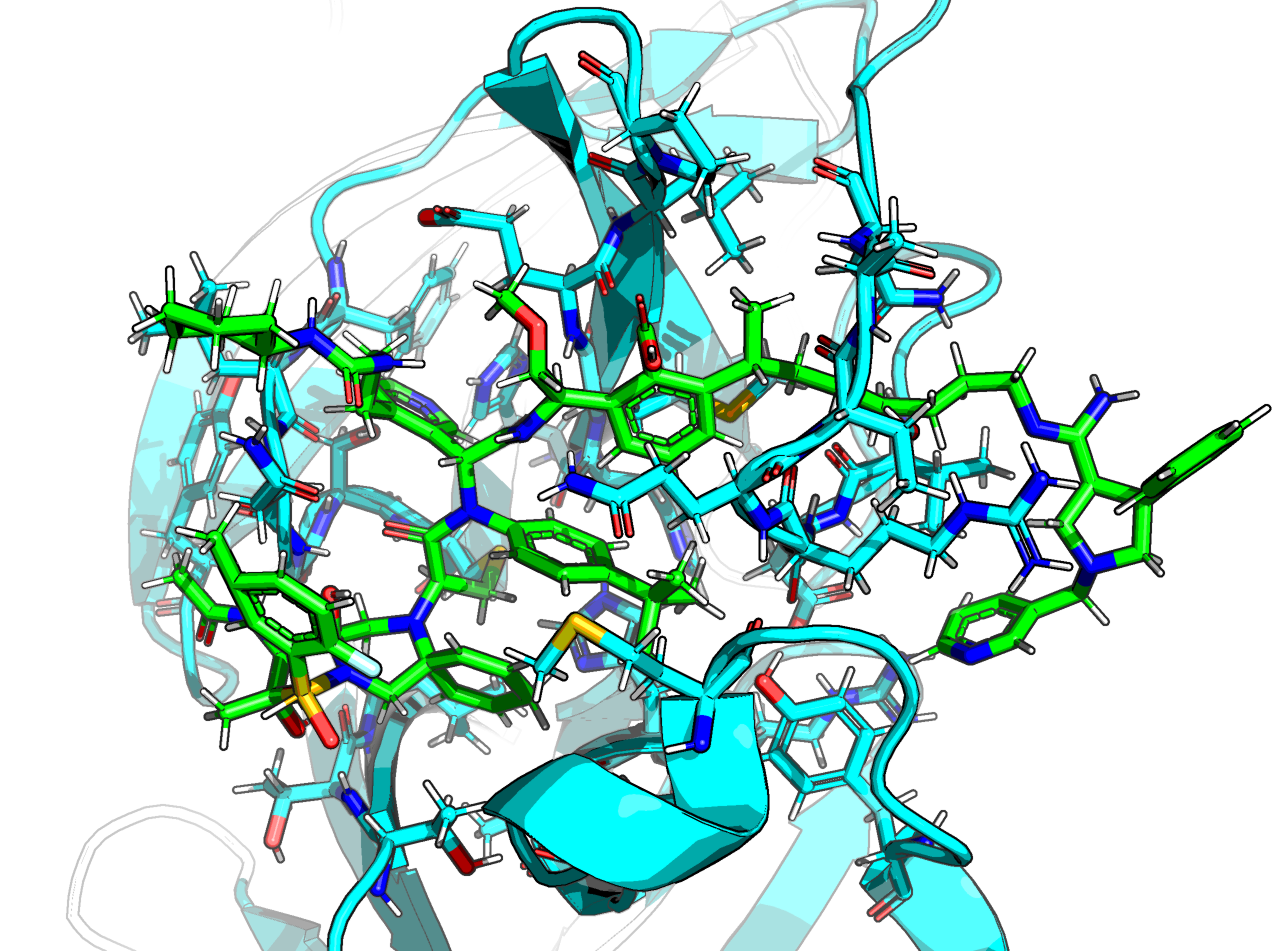
@frankvondelft, here is a interactive view: https://michelanglo.sgc.ox.ac.uk/data/ebd78eb8-6192-44ed-a38b-c822509a3179
The horrors are mostly from rings, but the oxycations to fix valence issues was my doing (makes sense only for nitrogen), in hindsight I should have taken the atom an atomic number before (X -> O -> N -> C). The ring downgrade from aromatic to single is probably a case not saving.
Comparing a 3D docked conformer for each hit from different programs (and the same programs with different methods) is fraught with problems. Obviously, there are an ensemble of docked conformers from every method and sometimes that ensemble is not very homogeneous - sometimes with molecules completely switched around adopting different conformers! If side-chains are allowed to relax (as they do), life becomes even more interesting!
I always favour using the biochemistry but usually there is little idea about which interactions are more important until a lead series is being optimised (and I’m often sceptical about any assumptions that there arent different lead series which interact in a different way).
This leads me to think about grouping hits in multidimensional space based on strength of residue interactions (where each dimension is a residue).
Just to clarify, Fragmenstein: assessing fidelty to hits is my “docked” set for the non-covalent call. This is a post about suggesting new followups with Fragmenstein (an algorithm that tries to place a stitched together compound from the hits and minimised it with strong constraints) — I submitted it to the wrong category.
Fragmenstein is not really a docking algorithm as its working backwards. This for exactly the reasons you said, namely different programs dock different and are complicated by a myriad factors. The XChem team did a fragment based search, so Fragmenstein aims to map the Moonshot suggested followup to the inspiration hits to see how good they were.
About flexibility of the protein. The minimiser is using pyrosetta, with backbone fixed but sidechains movable, in future I might merge the density maps of the hits in gemmi and use the combined density as a constraint and allow backbone movement when minimising the neighbourhood.
This is awesome! Do you have a .csv version of this file? I’d love to try to see how well the mergers re-dock.
That would be awesome.
data.csv (47.7 KB)
svg of merged (includes failed horrors) (1.7 MB)
zipped pickled pandas dataframe (requires RDKit) (209.9 KB)
Do note that the column exists means it did not crash and burn, not that it’s in Enamine REAL —I need to pre-filter for that as many compounds are impossible.
The set needs some work, as mentioned to @frankvondelft about the oxycations when a position gets a new bond it should shift back a tile in the periodic table and not be protonated. And I might repeat it with the glitchy ring collapsing code.
Matteo, I’ve finally had time to stare hard, and get excited: did you have time to do the “some work” you said it needs?
Because I’ve been remiss: we should move aggressively.
- Generate 3- and 4-way merges
- Submit them to Postera en masse so they can filter them for makability or availabiltiy.
Just a quick update. I fixed a few things in the code and made it do it’s thing.
The 4 way mergers are pretty impressive and there are less scary one —except for the outstanding issues mentioned below. The four way merger are bigger than @frankvondelft’s gigantic FRA-DIA-8640f307.
Outstanding issues: triangles from proximity bonding
The fixed included ring collapse to make ring mergers better —which it did (now there are no weird mega-rings.
The approach where the atoms remember what they bonded to caused some issues, so now when a ring is expanded back the atoms bond to their closest non-ring neighbour if with a cutoff. I need to do an ultra-simple fix to stop triangles from happening.
Outstanding issues: bridging selenium
The valance correction now does not add a charge resulting in oxinium ions, but shifts down the periodic table —seems to be fine, although there’s a bromide that becomes a selenium in one.
Outstanding issues: warhead mergers
The covalents stay colavent and are modelled as such, however, I need to make the code do a warhead change to make the merger work in the case of merging different warheads. Warhead changing is already in the code, so it’s straightforward.
Very cool…
But those triangles seem to dominate (x2908-x3110-x1311-x0771-x2705: 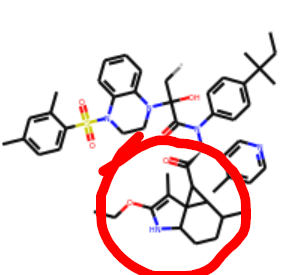
and it looks just as bad in 3D.
So I guess it’s only worth reviewing when it’s fixed - do you know when you’ll have managed?
I had fixed the triangle (and square problem) and a few others a few weekends ago and let it do its cycles with the silly ranking, which favoured mega-mergers. I also made it do more optimisation cycles as it struggles with larger “small” molecules (now takes 90 seconds per core (xeon e5-2640 v3) per molecule). I am uploading it now as a might as well as the ranking is silly.
The top scorer is over a kilodalton and crosses under a loop —it’s ∆∆G is a joke given its size.
There is less or very little dodgy chemistry, which was the aim.
Sorting by ligand efficiency reveals they aren’t that good — minus one kcal/mol per heavy atom would be a great result, whereas the best is -0.6 and 14 heavy atoms only (nice merger though).
For a less MW focused and more ligand efficiency focused approach, decomposing the fragments into sensible groups (rings, sulfates, amides etc.) would be a more sensible approach —and would simply be an extra pre-step. Although would add a fair amount of extra combinatorial complexity. RDKit has a dozen way to decompose a molecule and I have not played with them, so I would love to hear suggestions of which should be tried on the hits.
Woah hardcore… there’s lots of dodgy chemistry still in the top-ranked ones, though? Apart from the ridiculous size. We’ll have to figure out a way to whittle down… must mull.
Right at the top you have this: 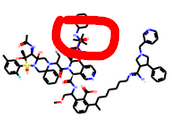
But yes, that’s a distraction… maybe just a size cutoff…
Uh-oh - use only fragments! I see you’re now using everything, but that includes Moonshot compounds, never mind where they bound (e.g. x2929).
The red dot means that it is on a different plane —RDKit unfortunately does not alter angles for the sake of visualisation unlike say ChemDraw. So the chemistry is normal just too much of it. The spiro rings are knowingly tolerated.
That makes sense. Especially as it is becoming a combinatorial suicide. And the size defeats the point.