The astonishing success of the Moonshot in harvesting designs from the community has left us with a new bottleneck: evaluating which designs best exploit the data, and are therefore worth making. (There is budget for ~1/5th of the submissions.)
The underlying premise of the whole effort is that the observed fragment-protein interactions are as close to optimal as possible, since fragments have so few interactions to satisfy. The corollary is that those compound designs will achieve potency that recapitulate precisely as many of these interactions as possible (i.e. same distance and angle) - preferably from multiple fragments.
So what we need, and what we don’t have, are algorithms/scores that will tell us that for each design, which were mostly generated (it seems) not by algorithm, but by people eyeballing the hits and using intuition to suggest how they might be merged or otherwise expanded. If a design merges several fragments while precisely recapitulating the various interactions, we need the score to rank it highly.
For example, JOO-PER-d7a-1_NIT2 may achieve this, at least when comparing its energy-minimized conformation (teal-green) to the positions of the inspiration fragments (grey sticks):
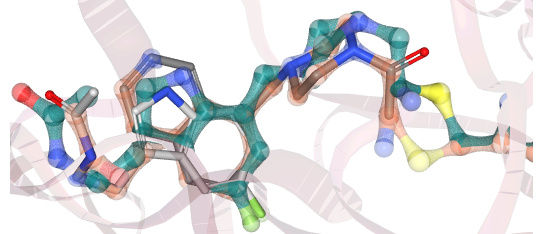
(Superposition generated by Matteo Ferla with this Fragemstein method, not yet documented.)
Existing scoring schemes seem to focus on extracting information from the hits about target engagement, e.g. for docking scores. However, this generates abstractions of the experimental data - which seems like a waste of precious data!
We now invite discussions and forum-post suggestions on how to do this. If you have a scheme already, by all means score the existing designs and submit them in the “Docking Results” category (the posts there have detailed instructions for how to format and upload them.): convincing rankings will be used to prioritise further compounds for synthetis and testing.
