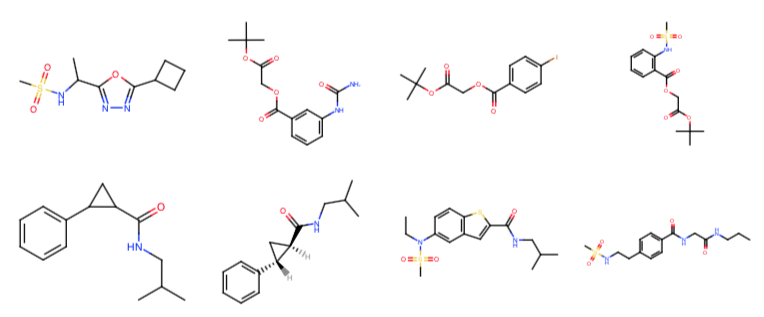
The Moonshot project has accumulated several synthetically accessible virtual libraries that would be very interesting to computationally screen for whether they might contain good “magic merges” of fragments from the X-ray fragment screen against Mpro.
It would be fantastic if the folks who have developed methods to look for fragment merges (@dmoustakas @matteoferla @tdudgeon and others) would be able to screen these datasets and report the top interesting hits:
Fragment Network expansions: @tdudgeon has enumerated compounds from Molport and Chemspace that contain the fragment hits as a substructure using the Fragment Network approach (code here). The largest set contains 85K compounds.
expanded-22_hac0-10_rac0-3_hops3.smi (2.6 MB)
London Ugi acrylamide library: @londonir has enumerated a library of Ugi reaction products accessible via parallel synthesis that possess an acrylamide warhead. There are ~39K compounds in this library.
ugi-enumeration.smi (3.2 MB)
London noncovalent pyridine-urea library: @londonir has enumerated a library of 15,279 non-covalent pyridine-ureas based on the fragment from x0434 that Enamine could make in parallel.
pyridine_urea.smi (623.4 KB)
London covalent piperadine-amide chloracetamide library: @londonir has enumerated a library of ~35K compounds for a covalent series for which there are three fragment structures (x1358, x1380, x1458).
piperidine-amides.smi (1.8 MB)
If any of these libraries contain high-quality fragment merges, it may be possible to rapidly synthesize some of them to test the fragment merge hypothesis.
2 Likes
@JohnChodera How are you proposing to do this? We start with SMILES so we have no 3D pose.
Are you suggesting to:
- do conformer generation to generate random conformers
- use docking to generate docked poses
- something else
p.s. I’m applying my “FeatureStein” approach to your docked poses and should have data on that soon.
@JohnChodera, @tdudgeon, I’ve downloaded these sets and are preparing them for docking into the single receptor model I posted about in the docking results forum. I’ll aim to post the docked poses for these sets sometime this weekend hopefully, if that will be useful for your analyses.
Cheers,
Demetri
My trail set is also available at Fragmenstein: merging
However, I have not filtered them against Enamine REAL.
I’ll run the other sets linked here in Fragmenstein to get a 3D pose and share the results.
@tdudgeon I used Fragmenstein to place and minimise the pyridine_urea set. https://www.well.ox.ac.uk/~matteo/pyridine_urea.sdf.zip.
Data:
pyridine_urea.xlsx (1.2 MB)
The template is template.pdb (370.3 KB) .
A lot of these clash with Q189, the neighbourhood was allowed to move but only sidechains, backbones movement was forbidden.
If these help I will do the other too!
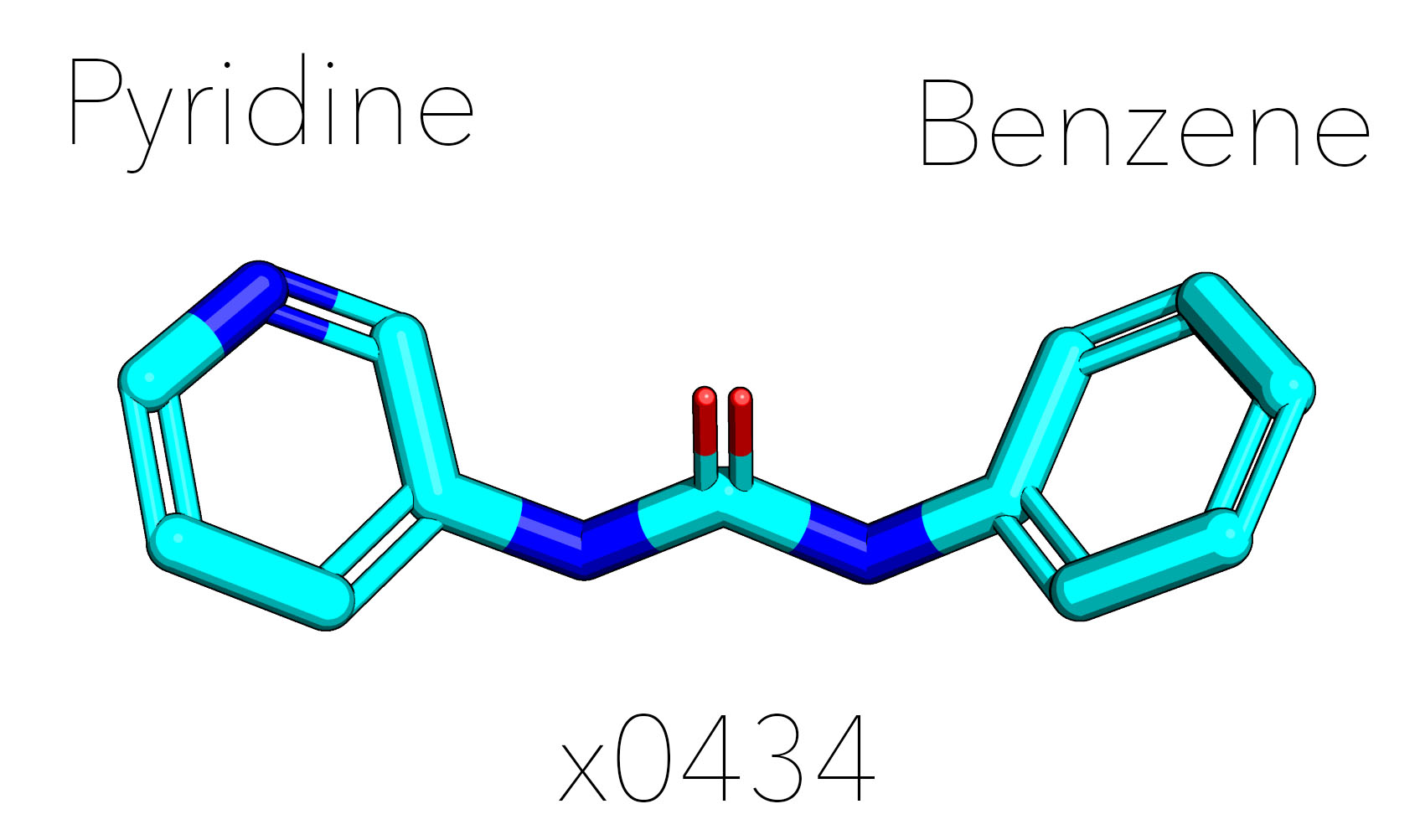
EDIT. The set was docked solely against x0434 as that was the hit mentioned in @JohnChodera 's post. This compound is nearly symmetrical (and technically can be fitted to the density either way):
As a result when you have either but not the same benzyl/pyridyl group, the preservered group will be placed. When you have a dipyridyl-urea core or a dibenzyl-urea core, then there is a problem as there would have been two equal solutions. The tie will have been broken based on atom index of the SMILES (in x0434, benzene is first, so reversed order for pyridine) instead of sampling both options as would have been more correct.
2 Likes
Here is the covalent set piperidine-amides, which have chloracetamides.
piperidine-amides.xlsx (2.1 MB)
https://www.well.ox.ac.uk/~matteo/piperidine-amides.sdf.zip
1 Like
In terms of fragment growing (i.e. using either or both linking and mergers), are quantifiable scores to compare across methods currently being used in COVID_Moonshot?
An example to illustrate my question.
Let’s say two methods are being compared for their ability to generate compounds from fragments A (IC50 = 1000 uM) and B (IC50 = 500 uM).
The top-scoring compound generated by Algorithm I is X, while the top-scoring compound generated by Algorithm II is Y.
When synthesised and tested, compound X has IC50 = 20 uM while compound Y has IC50 = 1 uM.
In this case, it might seem reasonable to conclude that Algorithm II outperforms Algorithm I, especially if that Algorithm II’s top ranked compounds consistently outperform those generated by Algorithm I.
Have any such quantitative measures been conducted or observed in this study? Currently, it seems that the efforts to merge the fragments mainly rely on docking studies, as opposed to using the quantitative data (e.g. IC50)–did I understand and interpret this correctly?
Many thanks,
Zhang-He
The docking and similar approaches as discussed in the category docking results were done to triage the human submissions to a reasonable number to order/synthesise and test.
Once we have/are starting to get more data in analyses to see which algorithm was best should and will be done —and I think @dmoustakas has done some work on the IC50 front already. However, from what I know, the data was not available for the initial hits — initial hits in FBDD are often un-assay-able biochemically with reasonable error bars.
From the crystallography only point of view, there is not only the boolean ‘binds?’, but there are per-hit corrected occupancy and per-atom B-factors. This data from the inspiration fragments could be useful for the ranking. I for one did not use this data as weights when placing fragments even if they could have very useful! The B-factors are present but would have required complex operations to normalise across the structures, while per-hit corrected occupancy depends on the solubility and other ligand-only properties, but I am told it’s a minefield not to enter.
1 Like
Thank you for directing me to the docking results category, @matteoferla!
I will have a look there to see if anyone has discussed docking results in relation to fragment merging.
can i get ic50 for these componds?
All the non-crystallographic data is held here: https://github.com/postera-ai/COVID_moonshot_submissions
However, only those that were chosen and have been synthesised will have an IC50.
The export CSV button in top right hand of https://postera.ai/covid/activity_data should also give you the latest IC50 info in case you don’t want to go via GitHub,