Dear all,
I just wanted to make a quick post detailing the design rationale for a series of structures that I will submit after this post. This is some work I’ve been doing since the UK lock down forced uni to shut. I am due to start my computational PhD in September so have treated this as a learning opportunity. I’ve been lurking on the forum for a while and can say it’s been very helpful in guiding my work!
I started by following a blog post by Pat Walters that builds upon a newly published method, CReM, which uses RDKit to suggest modifications to a given fragment by consulting a database of molecules and comparing them to the input molecule. This allowed me to generate a large amount of expanded compounds based on my chosen fragment, x0434.
I firstly identified positions on the fragment I wanted to expand and this was done mostly by eye and by overlapping the diamond fragments and seeing where on x0434 could I expanded to fill the pockets occupied by other fragments. For example, x0107 has a methyl group opposite the pyridine nitrogen and is also a reoccurring motif on this forum. Just as a 2nd example, x1249 has a nitrile group on the phenyl ring, meta to the urea, sticking into a pocket.

One input parameter into CReM is the maximum number of heavy atoms a modification can have. I set this at 8, which is somewhat arbitrary but it keeps the MW and heavy atom count down while allowing for the potential of 6 member rings with a couple of constituents.
As a result of these modifications a total of 7034 structures were generated.
all_expanded_smi.smi (433.0 KB)
As a first pass of eliminating some of these compounds, I again took inspiration from Pat Walters. In a follow up post to the structure generation, he takes the compounds and then generates multiple conformations for each compound within the binding site by mapping the compound to the core fragment. These conformers are then filtered based on two criteria:
-
Firstly, does the conformer clash with the protein? If yes, it is rejected.
-
Secondly, is the RMSD between two conformers below a cutoff? If yes, then one of the two is removed (i.e. removes what is essentially a duplicate)
This resulted in 2923 conformers from 1056 unique parent compounds (from the original 7034), implying that maybe my 8 heavy atoms was a bit excessive. The 3D coordinates of each conformer in the binding site are returned.
unique_parents.smi (38.6 KB)
Each conformer then goes through the following process to prepare for scoring:
- Protonation
- Small minimisation (To fix some ‘wacky’ conformations)
- PDB clean up
- Parameterised using Antechamber with AM1-BCC charges (to get prepi files)
- prmtops created using tleap with GAFF
- XML file created for easier use in OpenMM
- Conformer then added to x0434 crystal structure (with x0434 removed)
- Ligand and protein residues within 8A of the ligand were allowed to minimise (rest of protein kept fixed for efficiency)
- Ligand and protein then split into two pdb files
- pdbqt files created form the pdb files for use in AutoDock. AutoDocks prepare scripts were used for this.
Following that, each conformer was then scored using AutoDock Vina’s scoring function to score the resulting poses (Note I don’t actually use AutoDock to dock the compounds)*.
I’m not too interested in the numbers here because these scoring functions are not the reliable, but I am interested in the relative increases/decreases in predicted affinity compared to x0434. Below are the five best conformers for each position expanded (Note RDKit depicts the two meta substituents on the benzne as equivalent). For reference the predicted affinity for x0434 using this method is -5.30236 kcal / mol.

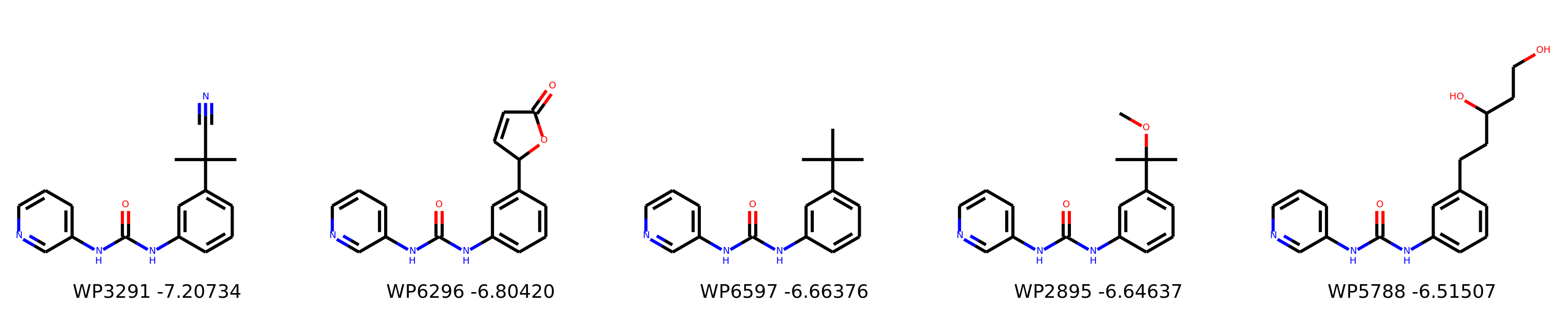
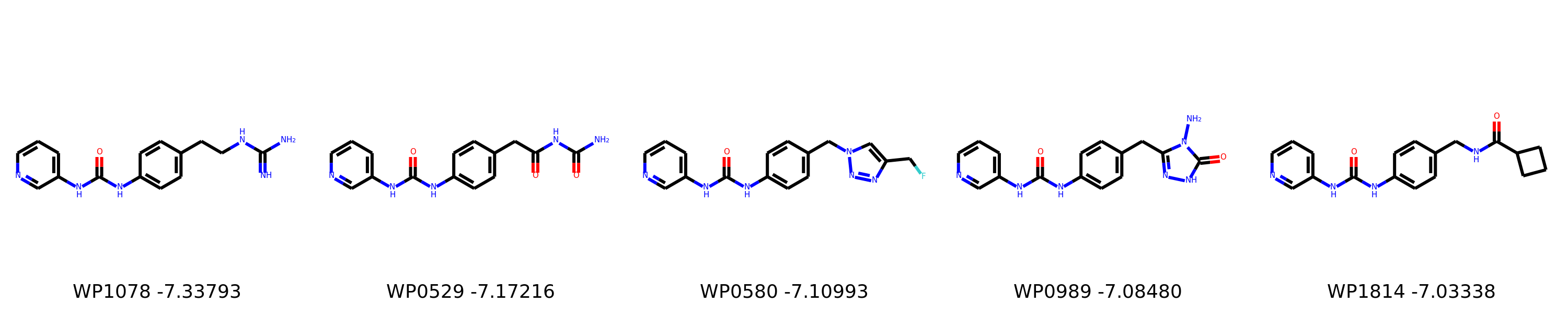
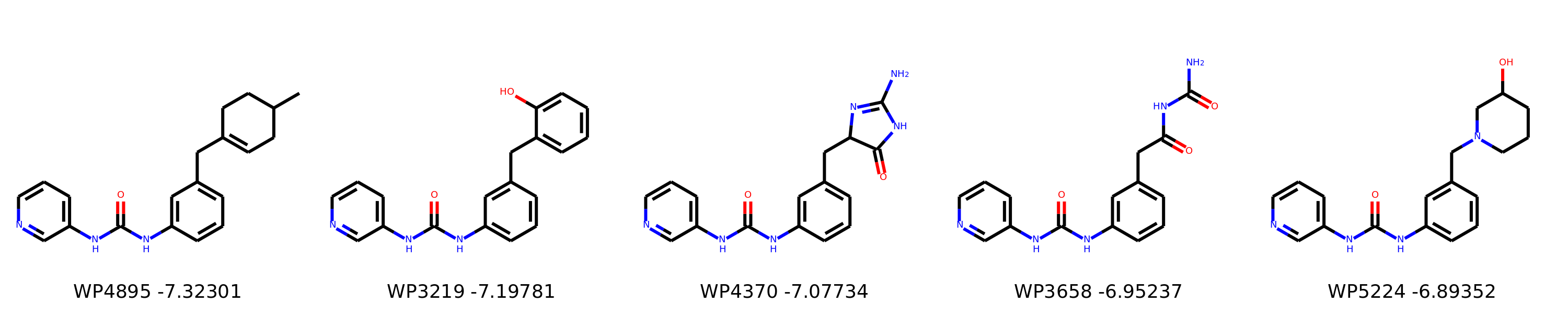
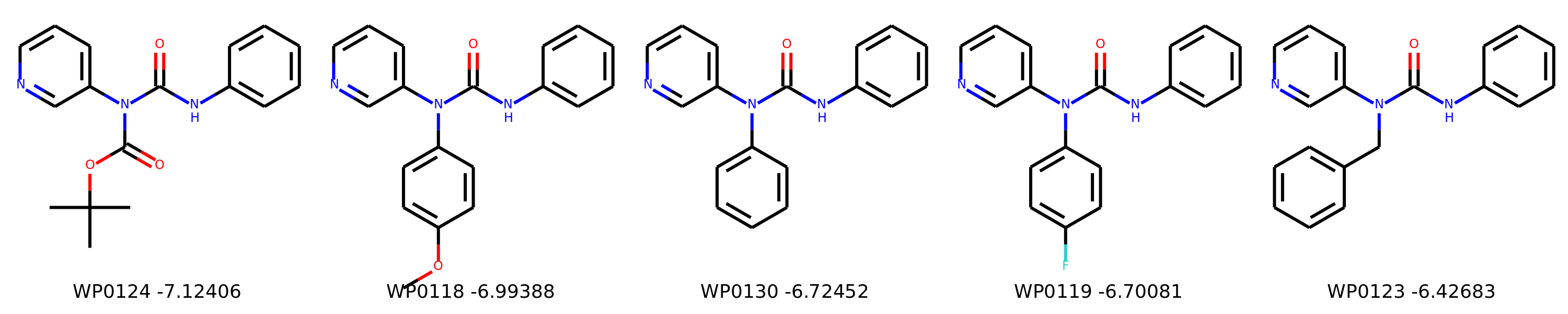

Finally, I combined a selection of these molecules and subjected them to the same protocol and scored them. Since I was combining 2/3 modifications, I repeated the protocol starting the final structures of both modifications and averaging the affinity.
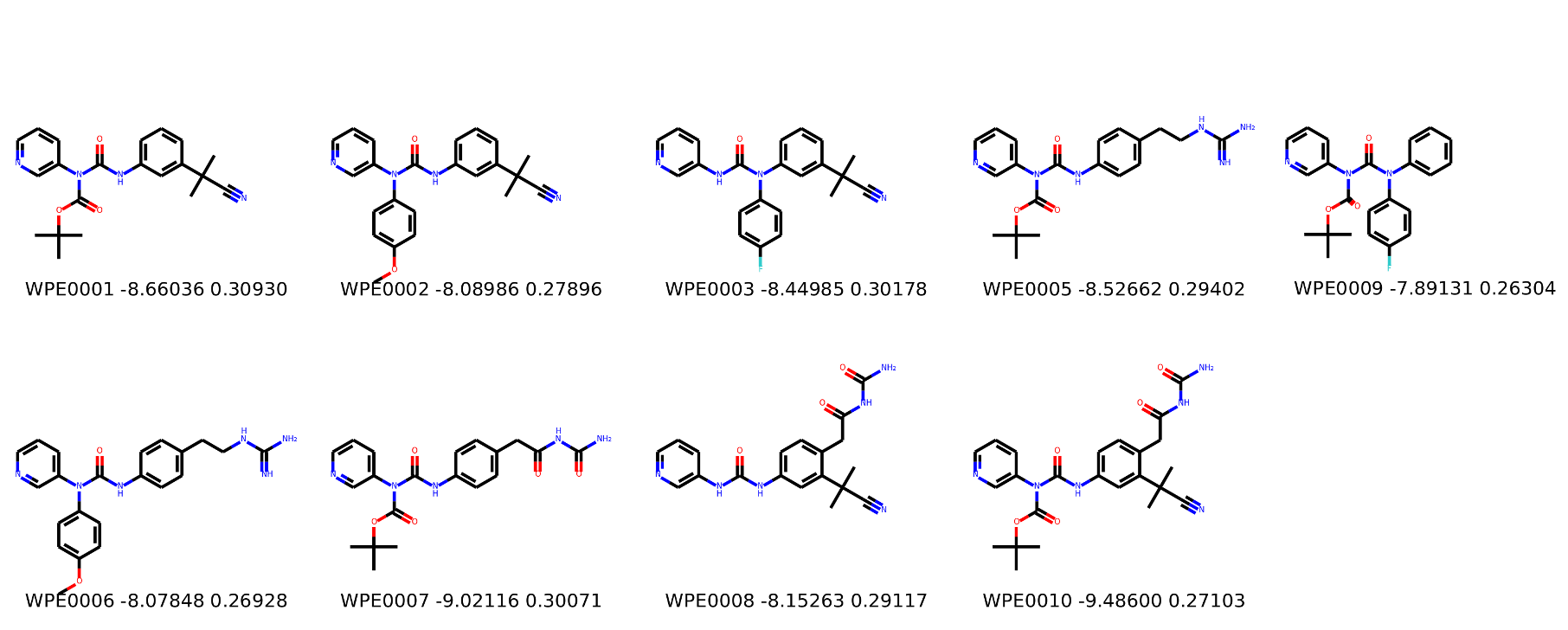
I think the main caveat of this is that generally scoring functions are biased towards higher MW compounds and as such some interesting compounds with a lower MW may have got lost in the list.
Full list of docking results here: docking_results.csv (56.7 KB)
I will upload all of these structures and hope this post was (atleast) an interesting read. I think it’s something a bit different!
*So why did I not just dock using AutoDock?
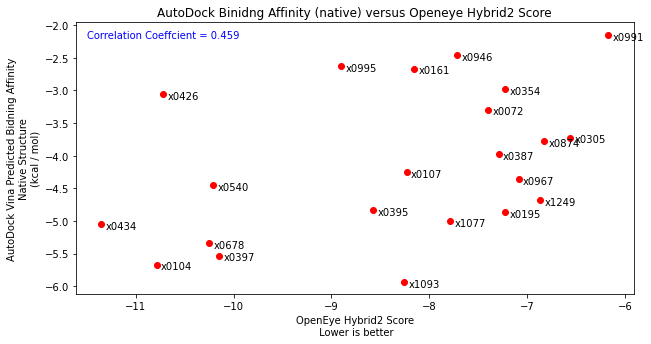
I had originally planned to do this, however after testing by docking the diamond fragments, I found that binding poses were not being well reproduced. So I then scored the individual binding poses using just the scoring function with the fragment in their native protein crystal structure. I compared these results with the docking performed by John Chodera with a correlation coefficient of 0.459. This isnt exactly perfect but also not horrendous (the relative rankings of fragments gives 0.5).
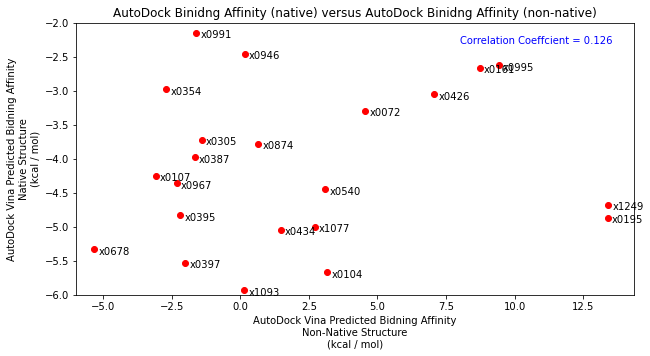
Following this, I tested the robustness of the scoring function by placing each fragment into the x0434 crystal structure (non-native structure). As expected, compared John’s docking and compared to the native structure results, there was no correlation highlighting the importance of having a native structure.
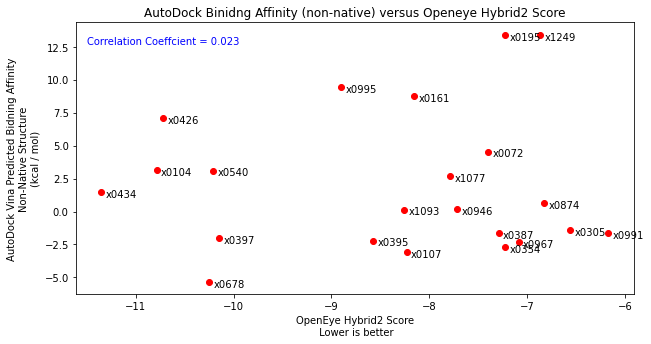
Finally, I then took each diamond fragment and subjected them to the protocol outlined above. Essentially placing the binding poses into the x0434 crystal structure and minimising the energy. Comparing the resulting structures by eye to their original crystal structure showed that the binding mode was well reproduced. Scoring these poses and comparing to the native structure also showed good agreement as well as improving the correlation with the Hybrid2 scores.
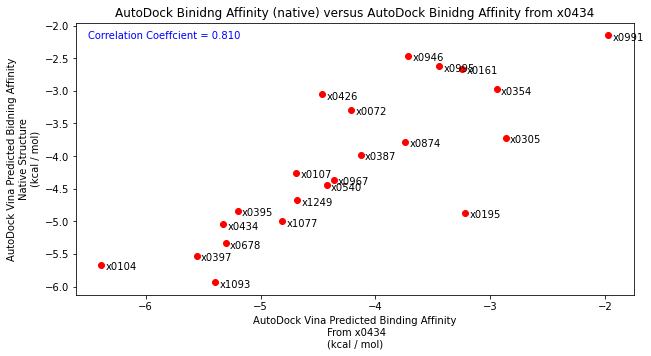
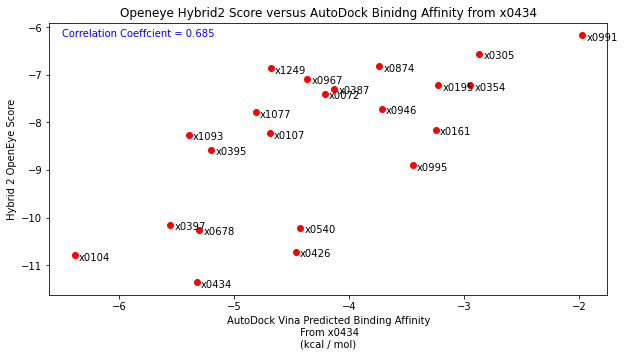
Anyway, if you’ve made it this far, thanks for reading! I would like to say thanks to everyone on the forum as well as a thanks to Pat Walters for his informative blog posts.