@londonir@mc-robinson
Here is the placeholder where you can upload the starting list for all compound scoring and docking.
List was prepared by @londonir with the following instructions:
Hi all,
For some reason, my constrained docking still results in some poses ending up with the warhead on the wrong side, and I’m not sure why—these poses should just be rejected.
The MD simulations examining the covalent warhead distance will be done in the morning, since I wasted time trying to figure out why the constrained docking was giving these surprising results.
Many well-ranked noncovalent complexes from (covalent_warhead_df-docked.sdf) do place the covalent warhead in a sensible geometry. These might be easily picked out by quick manual inspection of the top docking hits.
I’ve also sorted by scored overlap with the user-identified inspiration fragments at the suggestion of @frankvondelft (covalent_warhead_df-docked-overlap.sdf) and sometimes the geometries (see covalent_warhead_df-docked-overlap/) resemble existing fragments well,
but it’s hard to see good overlaps with multiple distinct fragments (as in @alphalee’s analysis) emerge organically in the top-ranked ligands. I probably need some other numerical criteria to figure that out.
The sensible geometries do look useful John, equally I like the overlap with distinct fragments, I appreciate all this is quite cutting edge - but I’d rather have this than just my medicinal chemistry biases. Even without a sortable score, if I can cut the list down with other means (crude size and lipophilicity will do) then I can eyeball docked structures.
This raises another point: designers might have made interesting merges of compounds by carefully looking at the angles, but not paid attention to the physico-chemical properties of the fragments they used for the merge, ending up with a greasy ball. The merge could be a smart one, but further tuning of the compound might be required, by an experienced medchemist. It feels like@JohnChodera’s overlap should be the first filter, and that a medchemist should review the list and maybe tune the merges that look the most promising to make sure that the compounds have good properties.
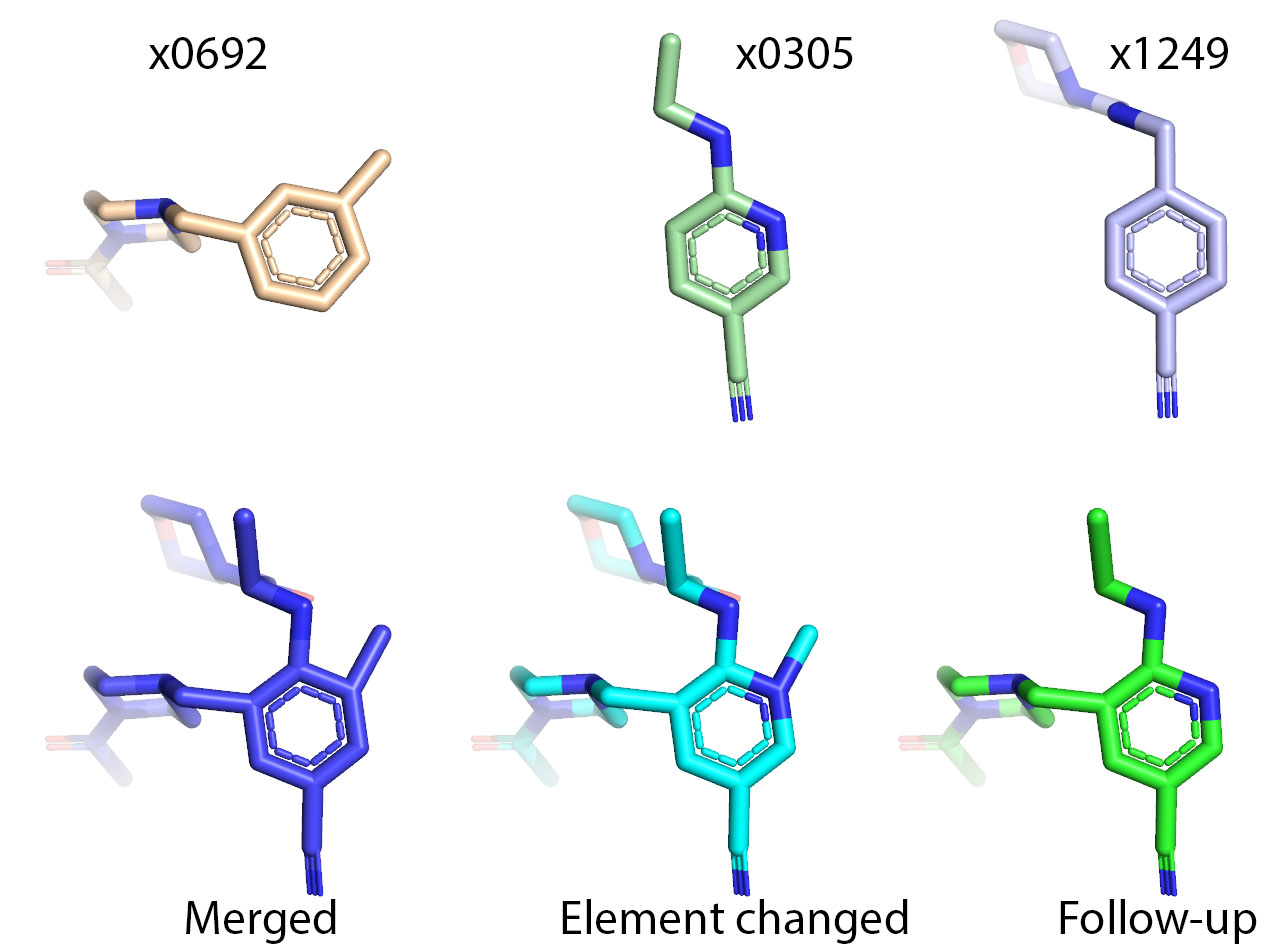
I have seen that some compounds are submitted with the basic amines protonated (they will be at physiological pH), others (including my submissions) are not. Before docking, are they all converted to the protonated form or can twice the same compound give entirely different docking results. There are a lot of piperazines submitted so this could have quite an impact on the docking outcomes.
I have completed the scoring of the covalent compounds follow-ups with strong constraints to a starting pose that is as faithful to the fragment hits
EDIT UPDATED LIST: fragmenstein_notes_v3.xlsx (286.8 KB)
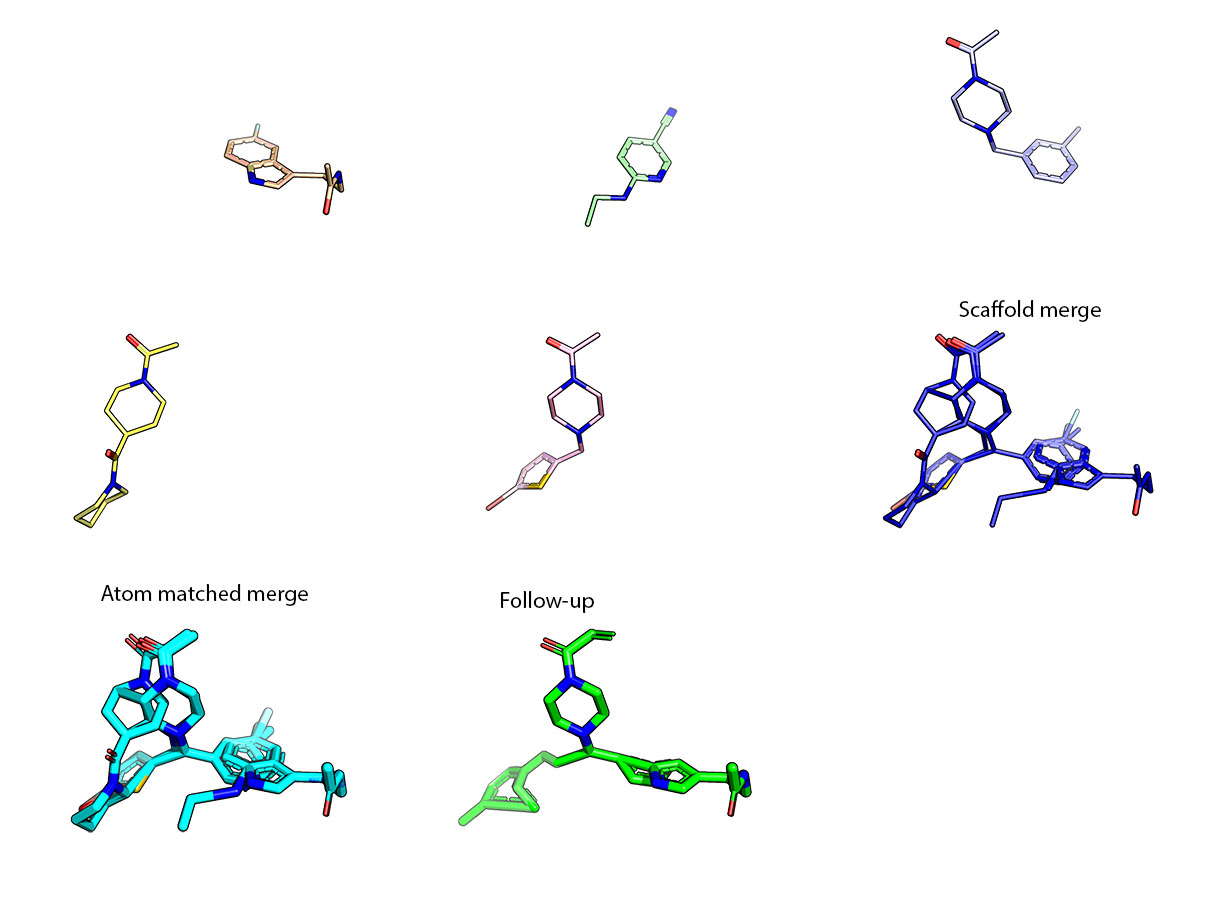
As discussed in the meeting, to make the starting pose match as faithfully to the hits, a collage of atom positions was made, a “Fragmentstein monster”. More details can be found at my github repo. If anyone wants the compounds for starting poses using other tools they can be found here.
Some look really good like this (often less than 3 starting hits):
The compound was not docked but was topologically minimised in the protein and scored (min1) and only subsequently it was docked like normal and scored (docked). Models can be found here.
An addendum to my previous post. The models and the data were incomplete, but I have fixed that. But only 870 compounds were able to be minimised. I will investigate into the why tomorrow or the Tuesday and make a compound only folder of mol files (with corrected bond order) in the in-protein minimised positions. Sorry about that!
Will depend on the other groups on the piperazines? If it’s an amide, urea, sulphonamide linkage coming off a piperazine N, it won’t be an issue but an H, alkyl/aryl group would have the N protonated.
if the piperazine ring is C-fluorinated you might find it’s weakly basic too and not protonated.
Continuous Process Improvement in the Manufacture of Carfilzomib, Part 1: Process Understanding and Improvements in the Commercial Route to Prepare the Epoxyketone Warhead
Peter K. Dornan *****
Travis Anthoine
Matthew G. Beaver
Guilong Charles Cheng
Dawn E. Cohen
Sheng Cui
William E. Lake
Neil F. Langille
Susan P. Lucas
Jenil Patel
William Powazinik IV
Scott W. Roberts
Chris Scardino
John L. Tucker
Simone Spada
Alicia Zeng
Shawn D. Walker
Cite this:Org. Process Res. Dev. 2020, 24, 4, 481-489
Hi @JHullaert! Apologies for the delay in getting back to you.
We use the OpenEye toolkit to enumerate all reasonable protonation states using oeproton.OEGetReasonableTautomers, and then assign protonation states appropriate to pH ~7 in solution. All the code to do the docking is here.
Note that this has limitations, in that it doesn’t account for the influence of the electrostatic environment of the protein on protonation states, and it does not make an attempt to penalize the the binding of different tautomeric/protonation states.
It does mean that different submissions that correspond to different protonation states or tautomers of the same compound should give identical results.
Our lab is working on automated sampling of protonation/tautomeric state Monte Carlo sampling within our MD-based free energy calculations, but due to technical constraints, this is still a little ways off.