I took a little break from coding to go back through the literature on SARS 2003 inhibitors, since the active site for MPro from the 2003 coronavirus and the 2019 coronavirus are very, very similar (with a few allosteric mutations that affect active-site binding…@RGlen is exploring this a bit).
One idea is that perhaps we can start with some of these inhibitors and test binding immediately on the SAR-CoV-2 Protease. If they are indeed still somehwat potent, then we can use information from fragments to help further build in potency / optimize the structure.
I have attached the following document with my cursory digging here: it focuses on small-molecules and SARS, not peptidic inhibitors or MERS – mostly due to time.
Happy for anyone else to contribute their insight. (I know @Chris.degraaf has already mentioned MERS inhibitors here Niclosamide SAR).
Perhaps most useful is the csv below of curated data. I have not curated every compound mentioned in the Google Doc (mostly because the manual curation was taking so long). However, I have automatically looked up if all of the compounds are available from Enamine/ Molport / MCule. We are planning to order many of these immediately to assay and get structures of from @frankvondelft 's lab
This paper below is quite interesting, because they show quite similar activity for their compounds across the whole corona family, and even beyond into the ‘super cluster’. It might be worth aggregating some of these more extended data for fragment re-splicing / machine learning efforts.
Kim, Y., Lovell, S., Tiew, K.-C., Mandadapu, S. R., Alliston, K. R., Battaile, K. P., … Chang, K.-O. (2012). Broad-Spectrum Antivirals against 3C or 3C-Like Proteases of Picornaviruses, Noroviruses, and Coronaviruses. Journal of Virology , 86 (21), 11754–11762. https://doi.org/10.1128/JVI.01348-12
My advice to the team would be to focus on reversible inhibitors (or at least on reversible inhibition as discussed below) and I believe that it will be necessary to exploit the catalytic cysteine thiol as a molecular recognition element in order to achieve the required level of affinity. In case it’s helpful, I have commented below on the some of the compounds in the spreadsheet.
A number of the compounds in the spreadsheet appear to have the potential to function as acylating agents. If the inhibition resulting from acylation of the catalytic is reversible then I think that the compound would be called a ‘suicide substrate’ although this is not an area of enzymology that I’m familiar with. Inhibition by acylation of the catalytic cysteine could also be reversible although I would not describe the inhibitor that behaved in this manner as reversible since it is cleaved by the enzyme. Acylating agents exploit the catalytic machinery of the enzyme and I would anticipate a degree of selectivity with respect to non-catalytic cysteines. IC50 values measured for acylating agents that inhibited reversibly would reflect kinetic factors (rather than affinity) and relating structure to activity is more difficult than for reversible inhibitors. This could be a viable way forward although design is likely to be more difficult than for reversible inhibitors.
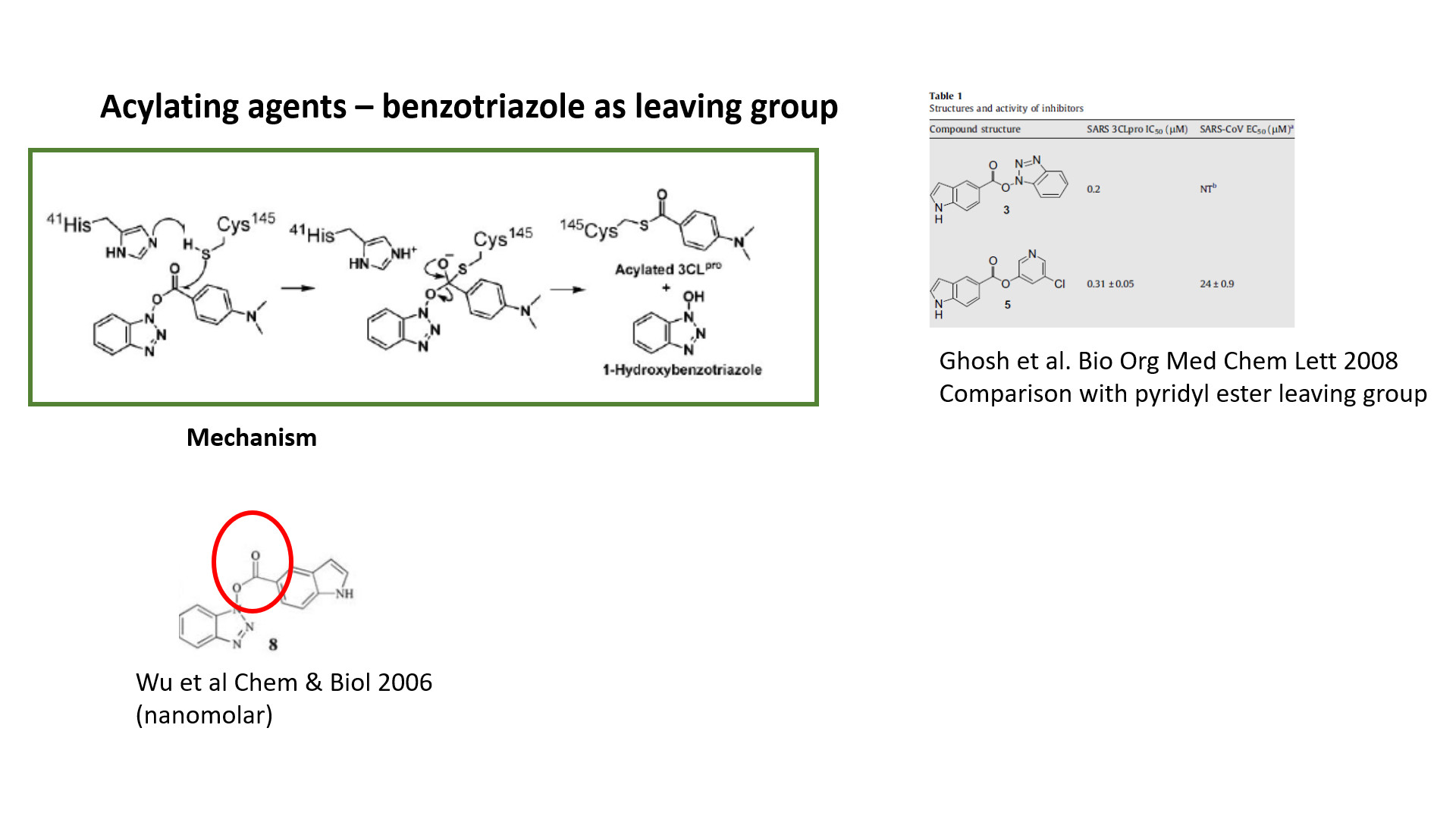
There are a number of compounds in the spreadsheet that I would expect to function as acylation agents. For example, aryl esters in which the aryl group is of an electron-withdrawing nature (e.g. pyridyl) or where the oxygen is linked to benzotriazole nitrogen. Also compounds in which a carbonyl carbon is linked to heteroaromatic nitrogen bonded to sulfur (especially when sulfur is also subjected to electron withdrawal by a heteroaromatic ring) or a nitrogen in a 5-membered heteroaromatic ring such as imidazole, pyrazole, triazole, etc. This article describes inhibitors of the latter type and, despite the claims of the authors that the inhibitors are non-covalent, I believe that the bound species is actually covalently-bound thiocarbamate:
I noticed a number of compounds in which the alpha carbon of an amide is bound to benzotriazole nitrogen and the possibility should be considered that the benzotriazole can function as a leaving group (just as cysteine sulfur displaces chloride from a chloroacetamide).
@jarvist, this is a point that really hit me from the Science and Nature papers already published this year.
In the Science paper, the inhibitors showed very similar potency across the COVID-19, SARS 2003, and MERS main proteases. Then the Nature paper just last week mentioned that the MPro structures from 12 species had very similar active sites. Of course, one wonders if this situation would change rapidly given the selection pressure of an anti-viral that targets the protease.
However, it does raise the point that collecting data regarding inhibitors of all of these COV species is potentially very useful. I’ve only had time to do SARS 2003, and only the non-peptidic inhibitors – but I think there is use in looking in the literature for others.
Thanks @pwkenny, this is a very nice writeup – and definitely appreciate the comments on the list.
With a few of the compounds in the list, I fear you are right that the IC50 is not that useful, since it is basically all driven by the irreversible inhibition. Some of the papers report K_inact data while others do not – and obviously kinetics become very important if looking for a irreversible inhibitor. Although we don’t have a clear focus (yet), I’d say we favor reversible mostly for the same reasons as you. The first focus above all else in our view is potency – with the qualification that a low IC50 for irreversible inhibitors is not necessarily useful in and of itself.
For example, the whole paper https://doi.org/10.1016/j.bioorg.2008.01.001 is devoted to taking some IC50=60nm Pyridinyl ester irreversible inhibitors and developing them into IC50=13-57 um reversible inhibitors.
Hi Guys,
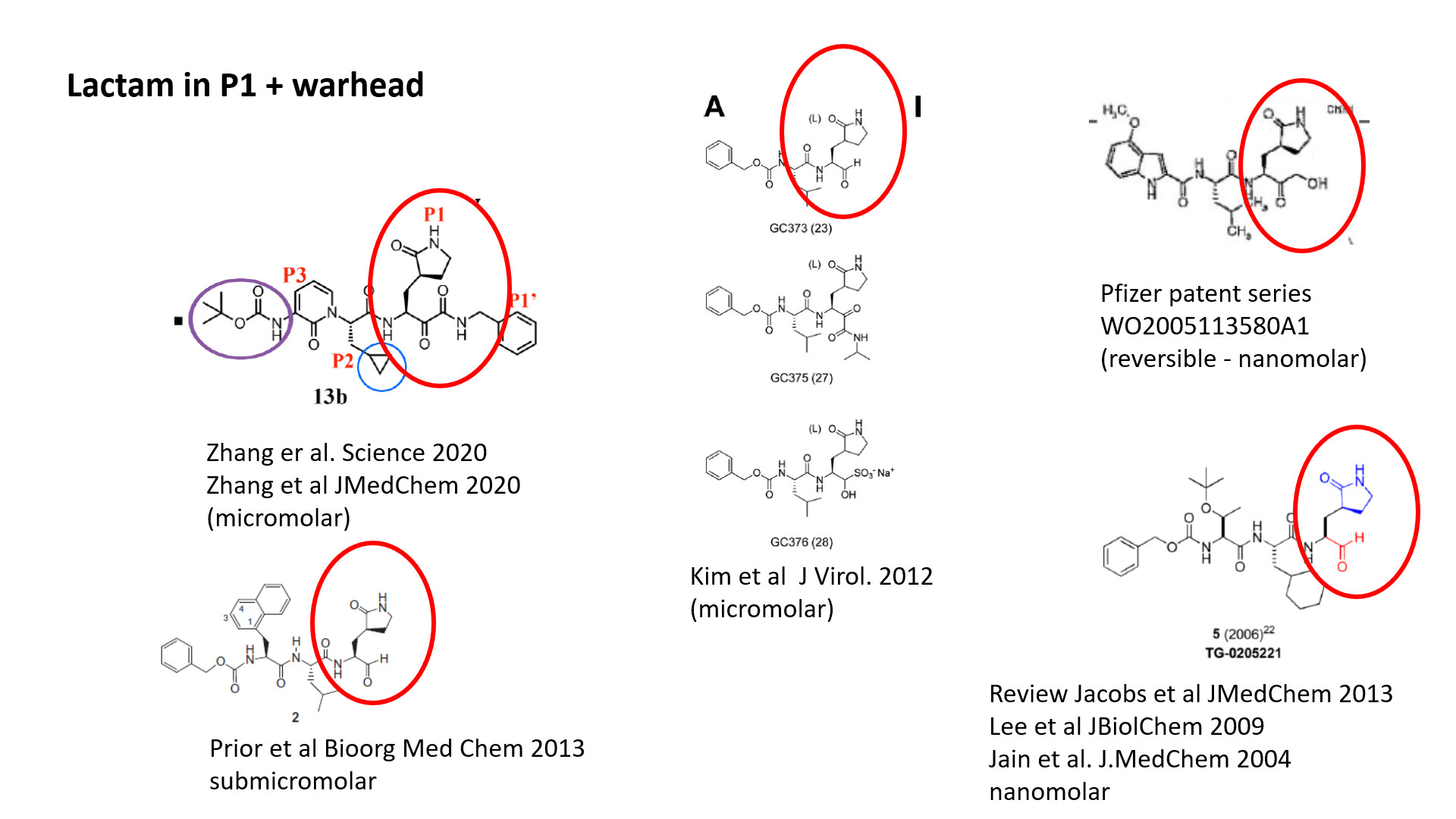
Thanks @mc-robinson, @pwkenny@jarvist for the nice overview of series and comments on it. I think excellent idea to see if some of these can be measured in the biochemical assays. To me it was quite overwhelming, so I made a few slides of the diverse series as they have been mentioned above (and one or two more). I grouped them in 4 classes.
Lactam in P1: these are quite promising and can be dressed with drug-like warhead such as ketoamides, although at the price of potency loss. The ketoamide warhead (from serine proteases) is probably not optimal for this protease, otherwise the potency would be better, I expect. The Pfizer non-covalent warhead, if it is one, on the other hand seems quite powerful, though this will need to be remeasured to be sure this is sound. The challenge here, I guess is to make these inhibs. less peptidometic to give them better PK and cellular activity, perhaps by replacing P2 and P3 with moieties from the fragments. It would also be worthwhile to explore better warheads, maybe learning from Cys-targeting kinase inhibitors such as ibrutinib and acalabrutinib (as discussed elsewhere on the forum).
& 3 Acylating agents (pyridyl esters and benzotriazoles). These can be quite potent biochemically, but I understand from the literature that they are not very potent in cellular antiviral assays. Needs to be addressed otherwise they are a dead-end.
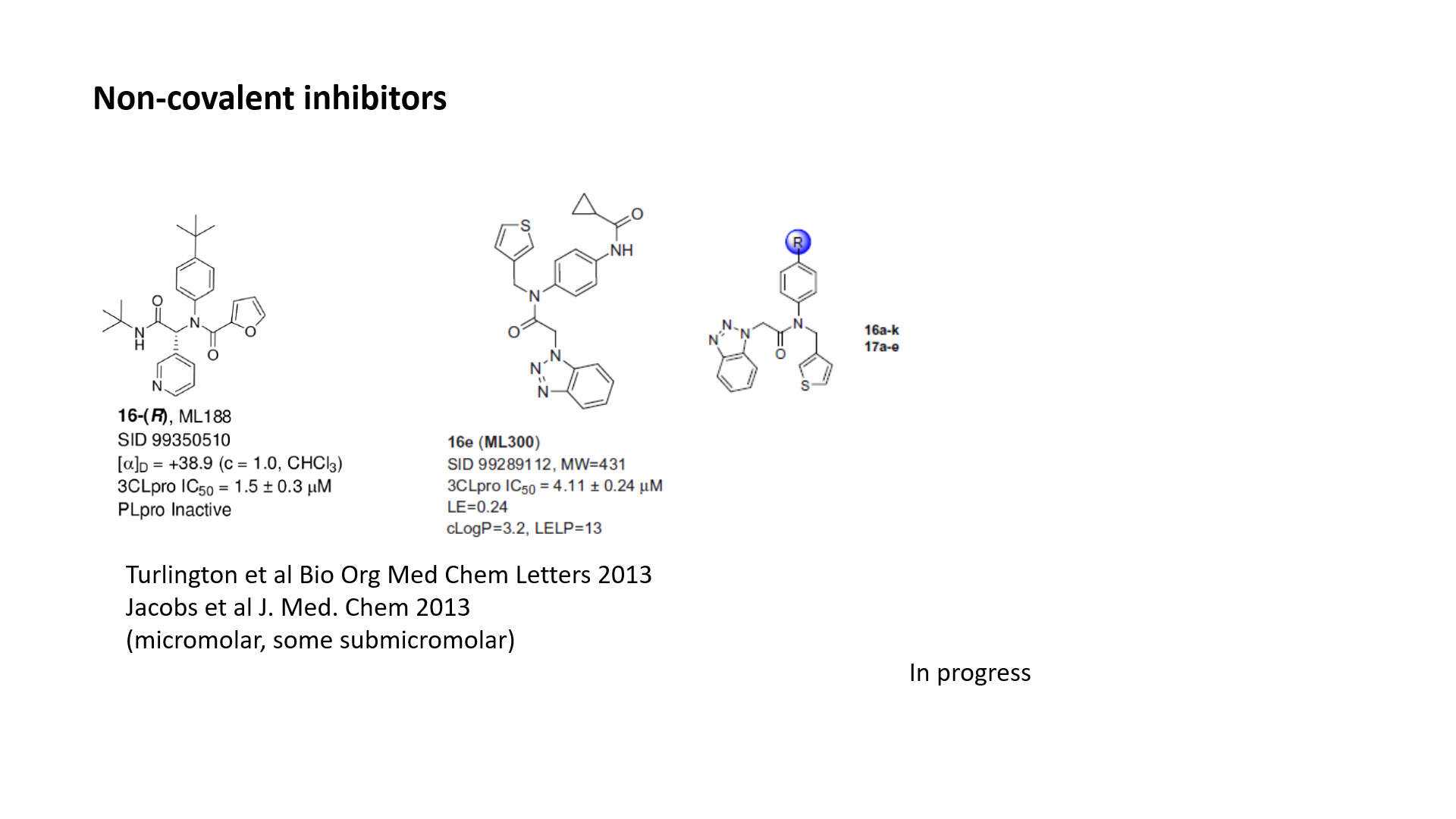
Non-covalent inhibitors. Particularly ML188 and ML300 would be interesting. Moderately potent, but maybe they can be improved helped by the fragments. Their potency might translate better towards cellular activity than the acylating inhibitors.
(References are easy to find above and on the web, I’m not sure if I can post the PDFs here)
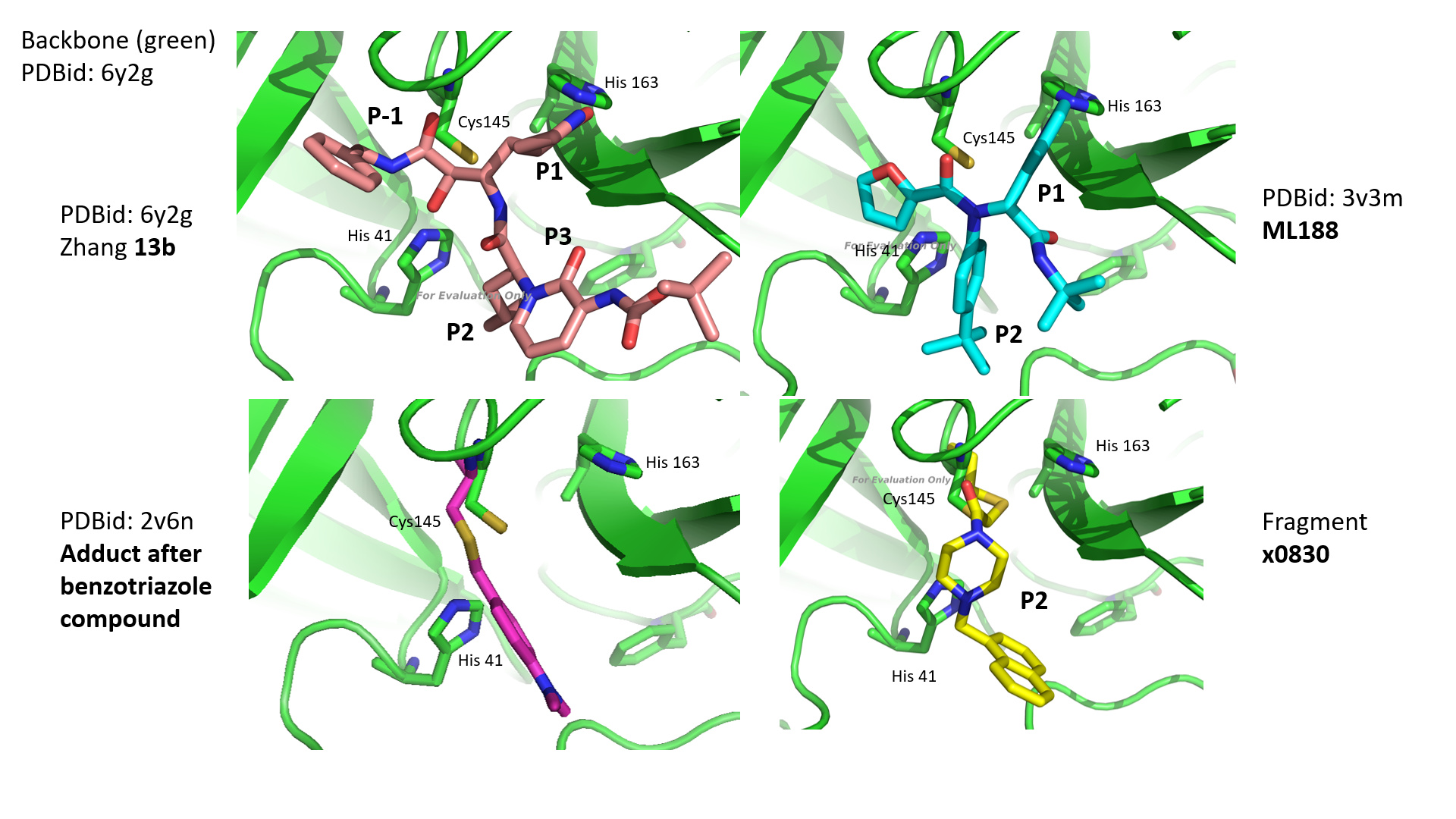
And here are pictures of the inhibitors from the various classes and how they fit into the active site. The viewpoint is identical for all frames. You can see that Zhang’s 13b ketoamide does quite a good job of spanning the active site. Also ML188. For comparison I included one fragment: x0830, not doing a bad job either.
Also nice to see, from a different viewpoint you see the general principles governing binding of the inhibitors. Both Zhang’s 13b and fragment x0830 present an amide-oxygen to the so-called oxy-anion hole, which plays an important role stabilizing the reaction intermediate during normal function.
You can also see that, as Zhang et al indicate, that the keto-group of the ketoamide manages to bind His 41 at the same time as the oxy-anion hole.
Fragment x0830 binds covalently in combination with the P2 site, skipping P1 altogether.
Stopping now. But lots of inspiration here, I think. Have a nice evening all!
I’ll first flag up a typo in my earlier post (I meant to say that I thought the term ‘suicide substrate’ applies when inhibition is irreversible). I do not think that it makes any sense to replace oxygen in the pyridyl esters with carbon if they are functioning as acylating agents.
Design objectives for acylating agents are primarily to make the transition state for the acylation step low and the transition state for the hydrolysis of the acylated species high. The transition state for the acylation step could be partly stabilized by non-covalent interactions between the leaving group and the protein (potential for tuning selectivity). While I’d expect design of acylating agents to be more difficult than for reversible inhibitors, we could think of using computational methods to generate models of the relevant transition states.
There are things that we could think of doing right now with the more potent acylating agents. For example, soaking experiments may allow the acylated cysteine to be observed crystallographically. It would also be useful to evaluate performance of a selection of compounds in the cell-based assays. Stability of acylating agents may be an issue and would need to be investigated.
I think the activity across the different SARS proteases is very encouraging since we might anticipate that other SARS proteases will need to be inhibited in the future.
Here is some more SAR that might be of interest:
Dipeptide aldehydes tend to be significantly more potent that the corresponding dipeptide nitriles. Azadipeptide nitriles are very potent when compared to the corresponding dipeptide nitriles (I think this because the adducts formed from the azadipeptide nitriles are more basic although I wouldn’t claim to fully understand this). Here is an article from a group (Nequimed) in Brazil that I work with:
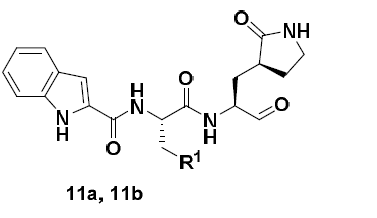
@pwkenny Very interesting. The 11a/11b from the Dai et al paper look like the ‘Pfizer-patent inhibitors’ above. They have nice potency with their 40-50 nM IC50 (although this doesn’t count for covalents, I know). It would be interesting to replace the aldehyde with a nitrile (or aza-nitrile), as mentioned in the Silva et al warhead paper you quote. Guessing from the x-ray structures, you would have to replace C=O of the aldehyde directly with C=N. If potency is good, the next step would be to make it less peptidomimetic. Actually you would be in the same starting point as the inventors of odanacatib when they started, as in the paper below (and that drug made it to Phase III)
Converting a peptidomimetic nitrile-based CatK inhibitor into a Phase III drug (odanacatib), https://pubs.acs.org/doi/abs/10.1021/jm0301078
(guess I’m getting off topic here -sorry for that)
Wow @JoostU and @pwkenny, this is amazing. This should become a review article!
Thanks especially @JoostU for making my info much more presentable, and for providing some nice PDB views.
I have some more reading to do based on your suggestions, but I think one question to put to you two and the crowd is what molecules of these do you think should be immediately made and re-tested?
We have also considered making many “matched pairs” of warheads on each scaffold to test the warheads. And additionally, we would likely look to get these inhibitors into crystal structures.
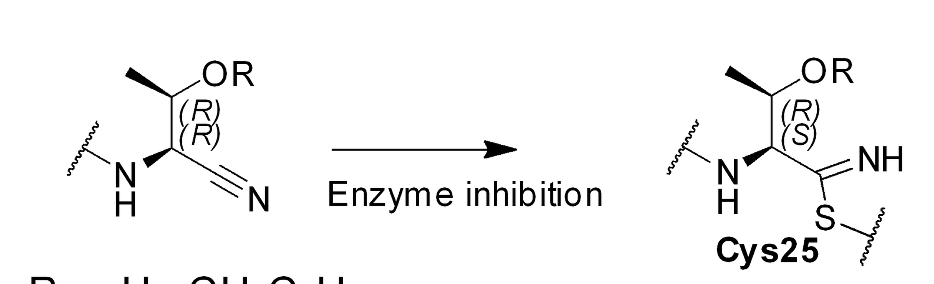
Just wanted to flag up a couple of points. If a covalent inhibitor binds reversibly then Kd can be measured and IC50 and Ki can be interpreted in the convential manner. It is even possible to use ITC to study the binding of covalent inhibitors that bind reversibly.
The hydrogen bond donor is not eliminated in odanacatib but the amide in question is replaced with an amine that is of low basicity on account of the CF3 group. However, the hydrogen bond donor of that amide can be eliminated by using the cyclohexane dicarboxamide scaffold:
Thanks Peter for pointing that out. I was too quick with my remark about Kd. I just posted the amide-aryl replacement picture as it was a first step in something that was quite a long journey by Merck Frosst to generate odanacatib from a peptidic inhibitor. In my time at MSD, I got to work on the project a few years and was really impressed. Quite a waste that such a good inhibitor got archived. Merck Frosst also had to solve quite some issues that only popped later in more advanced biological models. I think there is a lot to learn from this drug discovery trajectory for the Covid project. Here are a few more links, which I’m sure you are aware of https://pubs.acs.org/doi/abs/10.1021/jm800610j https://pubs.acs.org/doi/abs/10.1021/jm058198r
There certainly is a diversity of scaffolds, even when looking just at cathepsin K, and there could be value in seeing how some of the different scaffolds might fit into the SARS-Cov-2 protease. Pharma/biotech companies, who have been making noises about wanting to help, have large amounts of proprietary data in their databases and samples in their corporate collections.
@mc-robinson: I’ve docked this list using the OpenEye OEDocking noncovalent ensemble hybrid docking, using the bound fragments to pose these inhibitors in all fragment structures. All data is here:
The SDF file that @Waztom and @reskyner may be interested in is downloadable here. This isn’t yet in the new format since I think the SDF fields haven’t yet converged on specific tags and specifiers yet, but I’m happy to convert to a more convenient format when needed!
I finally got around to taking a look at past SARS inhibitor compounds that are not directly purchasable – to see if anything was in Enamine REAL or WuXi Galaxi close to the old compounds (Thanks to @Franca for the BioSolveIT license!) . I’ve collected that data in the large pdf here (sorry for the weird format, originally the document was a lot of code with plots mixed in). Do let me know if you see anything worth ordering. I have circled compounds I think may be worth ordering, but that was a very quick read over right before I got back to coding – and I would appreciate your input.
@JohnChodera also pointed out that the anilide inhibitors in this paper (see graphical abstract) have many similar compounds in REAL (including the compound below itself).
Some look quite lipophilic and maybe PAINs (Cl-nitroaryl group) but possibly worth a punt if a few can be readily synthesised. I notice, in the Nature paper, detergent assays were used to rule out aggregation effects. If these are purchased, is it worth rerunning to rule out aggregation effects?
(I started writing this a week ago, but forgot to come back to the browser tab + finish it!)
The 3CL-protease is well conserved between different viruses. Most of the active drug-like molecules are directly derived from the natural substrates. The best work I found on looking at QM/MM interactions with the substrate are the work of Paasche, who spent a PhD with Bernd Engels looking at SARS-CoV(-1).
The paper is probably the most dense high-level collection of their work.
Paasche, A., Zipper, A., Schäfer, S., Ziebuhr, J., Schirmeister, T., Engels, B. (2014). Evidence for Substrate Binding-Induced Zwitterion Formation in the Catalytic Cys-His Dyad of the SARS-CoV Main Protease. Biochemistry, 53(37), 5930–5946. https://doi.org/10.1021/bi400604t
But the details are better explained in his (freely available as a PDF below) thesis:
The thesis is probably also a good introduction to covalent inhibitors for this target. (For instance, see table 2-2, which lists all inhibitors as of 2013, with reference, PDB code, and activity.)