A lot of the ordered compounds are very interesting and diverse. My comment was fueled by the surprise to see a very strong focus on close analogues of the simple urea fragments in the list of ordered compounds.
My point of view is probably too simplistic for this large project and as non-expert in fragment based discovery I should be more carefull giving my opinion on things.
@JHullaert , please keep giving feedback – the point of this type of open collaboration is not to be shy about giving advice/ asking questions. We are all learning and adjusting as we go. I for example, was all ready to triage about 50-100 submitted designs, but then rapidly had to adjust cloud resources and change strategy to deal with the 3500 submissions.
Dear organizer,
Here come my top10 submission.
From all the submitted compound, I performed :
Molecular descriptors are calculated in order to visualize the chemical space based on druggable phys-chem properties (Hbond-don, Acc, NRotBond…).
Molecular fingerprints are calculated with obabel and compared to the other in order to build a NxN Tanimoto matrix used to visualize the chemical diversity of the submitted molecules.
Visual selection of several compounds pre-selected with the help of Phys-Chem and Chemistry spaces. (Using my personal submission as reference, and also trying to select a set of molecules that explore different part of the chemical space).
At the end I selected 3 covalent and 7 non covalent ligand.
Hi Lyna. I did some work on the PDBs with the ketoamide inhibitors. They nicely overlay with some fragments. See JOO-IND-926. And the screenshots I posted on the entry at the forum - best Joost
Top10.csv (620 Bytes)
Attached are my top 10.
I have excluded compounds with unprotected amides as although they are key for recognition by the protease they suffer from significant metabolic instability especially when dosed orally. This could be fixed in lead optimization but could also lead to significant loss of activity. Use of bioisosteres or steric shielding would likely provide metabolic stability.
I also excluded compounds with likely high lipophilicity/lipophobicity.
Some of the compounds possess significant pi-electron systems that would likely interfere with fluorometric assay, applying a PAINS screen would be highly recommended.
Thanks JoostU for your response. For keta-amide warhead, based on Hilgenfeld group’s extensive SAR, it is best to keep lactam at P1 and a small leu or cyclopropyl group for P2. So a fast and promising route to improve IC50 from micromolar to nM is to optimize P1’ and P3 sites. This design idea can take full advantage of crystalized covalent fragments as many of them overlay with the keto-amide warhead and P1’ benzyl in the x-ray complex. In addition, we also screened 470 drug fragments at P1’ and P3 sites using SILCS. Thus the designed molecules are quite big and complex because peptidomimetics do not necessarily follow the rule of 5 YUN-WES-64c. Appreciate any feedback. Thanks!
Hi Lyna. Your submissions don’t load on the forum page, so I cannot comment on them. Maybe @mc-robinson can help? I agree with you we should try to go for nM for these covalent inhibitors, and make use of a drug-like warhead like keto-amide. As you say, the designs are quite large and peptidomimetic and I think that will need to be addressed sooner or later. But it can be a start. Odanacatib (CatK cys-protease inhibitor that went into Phase III) is also based on a peptide that was converted into a small molecule by chemical design and synthesis cycles. On the other hand, one could also try to build out the molecule step by step, by attaching more and more fragments to a good warhead, check out X-ray, and improve again. Which I think is the route I would take, because it avoids shots in the dark. Although, if one of your designs turn out to be nanomolar, that would be a huge breakthrough.
A general question I have. Is there any perspective on getting Major funding to make more than 15 molecules? It seems such a waste of all these design to just make 15 scaffolds. I actually think EMBL or MRC should have a unit of 30 scientists working on this, funded by some economic relief package. What do you think?
Hi @JoostU, about off to bed, so I will fix error with @Lyna’s comments tomorrow morning and comment further on all of your great insights. But just want to note, we are planning to make far more than 15 molecules (sorry if that figure was confusing from an earlier note). We have around $300k of in-kind funding at this point and many some wonderful CRO’s working at-cost / pro-bono. We are also actively applying for more funding.
Having “grown up” without access to credible binding/modelling (GPCRs) in the early 2000s yet we progressed gastrin and PTH1 clin and preclinical candidates, I must admit modelling was never my first point of call. However, I’ve since gained an enormous respect and dependency on xray, modelling and calculations!
However, I feel we still need a human touch, lots of luck and not too much of an over-reliance on computational methods … We need a bit of everything. Our recent study showed that the order of predicted potency in a series of protein-Cys interacting fluoroalklyl groups was opposite to that actually found. We attributed this to a fine balance between protein binding and desolvation penalties. Are we looking at desolvation here also?
Dear JoostU, here is the pdf verison of the submitted alpha-ketoamides. Six Mpro-x covalent fragments were used as P1’ group because they perfectly align with the ketoamide P1’ benzyl group. covid-19-Mpro-design-Luo.pdf (48.5 KB)
Hi Lyna. Thanks. What I actually meant to type is that I couldn’t place my reaction to your molecules at the forum place associated with their entry, the place designated for reactions to designs. That’s why I put it here. Thanks for taking the trouble of attaching the .pdf - best Joost
Here is my selection. Chosen on the basis of covalent warhead, presumed correct length and orientation of groups, absence or minimum number of chiral centers, absence of toxicophoric groups, and industrial synthesis quite accessible.preferences.csv (493 Bytes)
@JSPEN I think you are totally right! Right now, I don’t think anybody really cares unless we get something quite potent… Though I will note that a couple of compounds in the literature showed a distressing drop-off between protease IC50 and anti-viral EC50 (so those other issues may come more quickly than we want them to)
One proposal for getting the potency early is to start with the past inhibitors of SARS and build in the fragment info. Overall, potency has transferred pretty well among other compounds (not a huge sample size though). More on this here A brief exploration of past SARS small-molecule inhibitors
Hi @JoostU and @Lyna, first, sorry about the error with your submissions not showing up on the forum. That should now be fixed.
And I appreciate both of you pointing out interesting overlaps of the fragments with the ketoamide inhibitors. I think the approach of combining known inhibitors with info from the fragments is a very interesting approach. It’s one that I think we are also considering with some non-peptidic compounds as well (mentioned here A brief exploration of past SARS small-molecule inhibitors).
The issue of peptidic vs non-peptidic is still an open one to be explored – and I am not sure I have a great answer yet as to which is the better route.
@JSPEN, interesting perspective – and agreed a lot fo help from a lot of areas (and luck) is needed. One interesting thing about this project is that we are seeing newer codes and approaches being tested prospectively on a short deadlines. This is very different from the usual retrospective tests we see with lots of times in papers. (Though yours is a nice exception)
You might want to get your own submission in before having a peek to prevent any peer bias.
Note that exactly ZERO of the submissions receiving votes from multiple reviewers are in the Wave 1 order, and only 10 of the 102 distinct submissions that were deemed “top” are in Wave 1. Perhaps an interesting datapoint in the debate about selection criteria that weigh manufacturability more heavily than perceived efficacy.
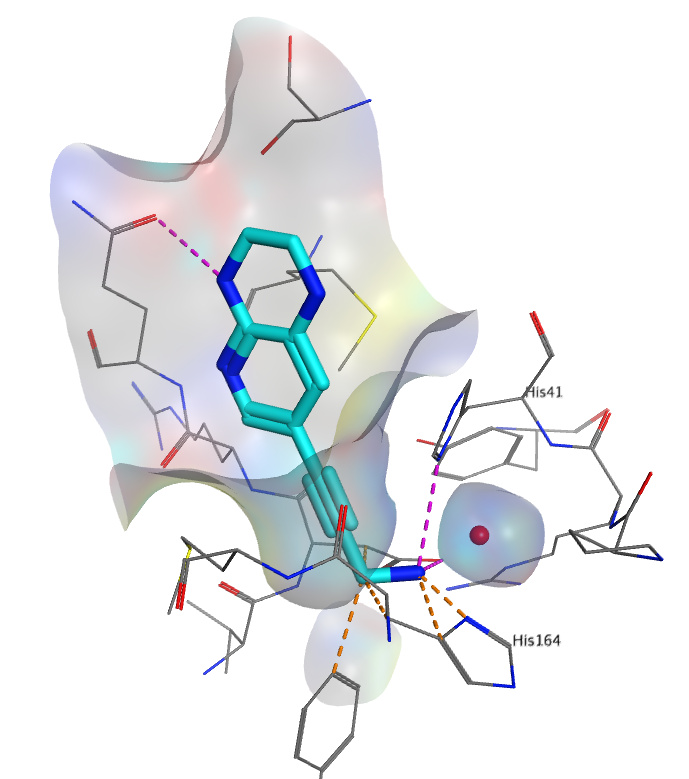
Hello everyone! Unfortunately, I found out about the project quite late. Nevertheless, I want to contribute a suggestion for your consideration. As I understand, it is different from what was suggested before. I looked at the crystal structures of non-covalently bound ligands and noticed that the one with cyano group goes deep into the pocket, maybe the straight conformation allows that. So my suggestion is to extend this part of the molecule to reach out to conserved water molecule and interact with it as well as with nearby histidines. Substituting of water molecule is risky, therefore, I would not start with that and add a small substituent. So this is my suggestion: n1cc(cc2NCCNc12)C#CCN
Similar compounds have been patented as anti-arthritis agents. The molecule would fit only with induced fit docking, but the water molecule has to get there somehow, so induced fit should be possible at this part of the pocket. Piperazine was added to constraint the molecule (might be entropic gain) and add H-bonds with waters. Picture (hopefully) attached.
Really appreciate you summarizing this @jamma and for your insightful comments. Will certainly bear this in mind as we move to the next selection criteria.