Topic automatically created for discussing the designs at:
https://covid.postera.ai/covid/submissions/EDJ-MED-41558c53
Hi @edgriffen placing a hydroxyl group at the periphery of the molecular structure does seem to be a rather unusual tactic for reducing metabolism given the potential for hydroxyl groups to get conjugated (e.g. glucuronidation). My understanding is that the potential Ames liability stems from the possibility of the P1 aminoheterocycle being liberated by amide hydrolysis (after which the entire P2 portion of the molecule is irrelevant). I may be missing something but I really don’t see the pendant hydroxyl as a solution to the problems that you’re attempting to address. In case it is of interest I’ll mention @mc-robinson @Ben_DNDi @alphalee who have also included hydroxyl groups in their designs.
We also have to get the metabolism in rat down further, compounds 12 and 13 have precedent for metabolism reduction in the MMPA literature, the quaternary centre removes the possibility for oxidation and presumably is slower for glucuronication or sulfation due to steric hindrance. We want to keep the hydrogen bond donor in place as that seems important from the enzyme SAR. Though looking at the crystal structure of MAT-POS-4223bc15-23 I still don’t completely understand where the potency is coming from XChem @ Diamond
Hi @edgriffen
Apologies for the length of the message.
Could you point me to the MMPA literature that supports the view that designs 12 and 13 are likely to lead to improved metabolic stability? Specifically, how were the MMPs defined and would the metabolic stability readout have registered conjugation of the hydroxyl?
My other worry about hydroxyls is permeability. I used MMPA at AZ around 2005 (if memory serves me correctly it was just before I escaped from the IGF project) to show that substituting aliphatic carbon with hydroxyl led to reduced passive permeability and increased efflux. While I’m certainly not suggesting that you should give weight to ancient MMPA for which I can’t even properly remember the details, it could be idea to re-visit this using Medchemica capability (you may already have done this) since the hydroxyls are showing up in a number of submissions. If you’re doing this, it’s really important to make sure that both members of each pair are from the same assay (there were results from both internal and external Caco-2 assays in the 2005 AZ database).
I wrote some notes on the MAT-POS-4223bc15-23 crystal structure which may be helpful. The pendant amide of MAT-POS-4223bc15-23 does not appear to make contact with the protein although there does appear to be some contact with the P1 isoquinoline. I see the contribution of the amide NH to the potency of MAT-POS-4223bc15-23 as resulting from its alignment with the tetrahydroquinoline nitrogen lone pair which stabilizes what is an ‘unusual’ orientation for a pendant group. Linking the pendant group though sulfonyl also places it in this ‘unusual’ orientation although for a different reason (sulfonamide conformational preference). There is also the question of a potential interaction between the amide NH and the carbonyl oxygen of the ‘central’ amide (I’ll mention @RGlen with whom I’ve discussed this previously in case he would like to join the discussion here) although I would expect the amide NH to align with tetrahydroisoquinoline nitrogen lone pair even in the absence of the ‘central’ amide.
There is a subtle but quite significant difference between the tetrahydroisoquinoline and dihydroisquinolone scaffolds with respect to the orientation of the pendant group. Specifically, what is the ‘unusual’ orientation for a tetrahydroisoquinoline becomes the ‘default’ orientation for the dihydroquinolone. One implication of this difference is that it would be unwise to automatically assume that the pendant group SAR will map directly from the tetrahydroisquinoline scaffold onto the dihydroquinolone scaffold.
Getting back to EDJ-MED-015fb6b4-2, I can see a 20 nM IC50 for the active enantiomer (CVD-0019230) in the CDD database. I would anticipate permeability problems for this inhibitor (there are two secondary amide groups as well as the dihydroisquinolone carbonyl oxygen) although I’d be very happy if this worry was shown to be groundless (i.e. good oral absorption with potent enzyme inhibition translating to potency in cell-based assay). If, however, you do experience permeability problems then these are likely to be difficult to fix and they will also constrain your options for addressing metabolic issues. I would expect EDJ-MED-015fb6b4-2 to bind in a similar manner to MAT-POS-4223bc15-23 and would expect the primary effect of elaboration from the pendant amide N-methyl to be addition of ballast.
I think that it would be worth considering replacements for the pendant amide of EDJ-MED-015fb6b4-2 especially with a view to eliminating the hydrogen bond donor (it’s important to not lose sight of amide hydrolysis as a potential clearance mechanism). The design team might wish to consider PET-UNK-c7ac4d9e-1, the ester analog of EDJ-MED-015fb6b4-2 as an easily-synthesized compound with the potential to inform (isosteric with respect to the amide, hydrogen bond donor is eliminated and polarity is reduced) design of pendant amide replacemements.
MAT-POS-a54ce14d-2, the active enantiomer of EDJ-MED-015fb6b4-2 has an EC50 of 70nM in the A549 cell assay, so permeability from a cell perspective isn’t an issue, however it’s still cleared too fast in the rat. Apologies for being a bit slow replying, we’ve some other issues to deal with. Worth looking at the latest batch of crystal structures: X-ray structure release - #11 by Daren_Fearon
Hi Ed
Did you look at the metabolism of [EDJ-MED-015fb6b4-2] by MS? Are you losing the NMe (could you replace with NiPr ?) or are the pyridines taking a hit? Happy to try the CHF2 Baran test if you want us to! CHF2 could then be tested for stability.
Hi PWK
What about a NMe2 to replace NHMe or morpholine/piperazine to lose the HBD in the amide?Might tackle efflux issues. Wouldn’t a Me ester be rather labile too? OiPr?
Hi @edgriffen, I agree that translation of potency in the enzyme inhibition assay to potency in the cell-based assay is strong evidence that permeability is not an issue for MAT-POS-a54ce14d-2 from the perspective of the cell-based assay. I can also see the oral PK data for MAT-POS-a54ce14d-2 with bioavailability in the range 6% - 7%. The low bioavailability could be due to first pass metabolism, poor permeability or a combination of the two (my understanding is that rat bioavailability tends to be lower than human bioavailability although I’ve not looked at this for over a decade).
Amide hydrolysis is still a potential issue and I would guess that the pendant amide of MAT-POS-a54ce14d-2 would be more vulnerable on the basis of being at the periphery of the molecular structure. I’ve submitted PET-UNK-f860268d-2 (des-methyl analog of MAT-POS-a54ce14d-2) since the primary amide may be a worse substrate for hydrolytic enzymes. @JSPEN has suggested the NMe2 analog and I think this would be worth looking at (while the HB donor in the pendant group is necessary when using the tetrahydoisoquinoline scaffold, it is unclear that this will still be the case for the dihydroisquinoline scaffold). I would anticipate that the [NHMe>>NMe2] modification of MAT-POS-a54ce14d-2 will still lead to a loss of potency (the pendant amide will probably need to rotate by 180° around the CH2-C(=O)N bond) but I’d expect this to less of an issue on a dihydroquinolone scaffold. While I think the [NHMe>>NH2] and [NHMe>>NMe2] analogs of MAT-POS-a54ce14d-2 would be worth including if you’re trying to solve the clearance problem by pendant amide variation, it would be prudent to also consider amide replacement options (e.g. with heterocycle).
Hydrolysis of the central would come with the additional problem that it generates something rather less wholesome than methylamine. The spiro ring fusion tactic should ensure that hydrolysis doesn’t happen (and that Ames active aromatic amines are not generated) but comes at the cost of increased synthetic complexity and likely loss of potency relative to non-spiro analog. Another option is to cap the central amide NH with cyano which I’d expect to not to invert the cis/trans geometrical preference of the central amide (a problem with methyl-capping anilides). I’d expect the capping cyano to be close to the catalytic cysteine and covalent bond formation, if it occurs, is likely to result in increased potency. I have submitted a number of designs which feature cyano-capped amides although I think the best way forward would be to first synthesize PET-UNK-5ecb6237-1 so that potency can be checked and synthetic issues can be identified.
I’ve checked the protein structure update but not seeing any dihydroquinolones.
Hi Pete,
Lots to reply to here, the spiro cyclisation does add an additional two steps, but they’re worked out now, so that’s not critical, I consider these two equipotent: 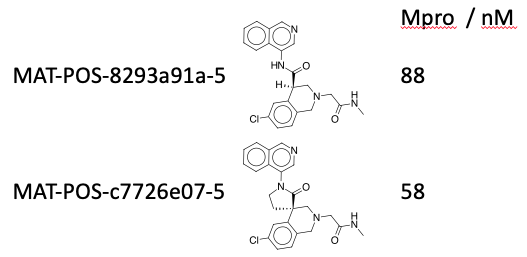 as purified single enantiomers - but the cell data is what matters.
as purified single enantiomers - but the cell data is what matters.
We have made two pendant glycine amides with C(=O)NMe2: RAL-THA-5e0a6f1a-1 (734nM) and MAT-POS-4223bc15-24(836nM) both considerably less potent than their mono methylated analogues EDJ-MED-015fb6b4-2 (16x more potent) and MAT-POS-4223bc15-23(14x more potent), so I’m more minded to keep the hydrogen bond donor in place, though interested in looking for ways of reducing the probable cleavage in vivo. We have some heterocyclic amide replacements in synthesis - more suggestions there would be welcome. EDJ-MED-12c4873b-2 is interesting as the cyclopropyl glycine would be expected to reduce nucleophilic attack on the carbonyl - and it retains potency.
Hi Ed,
I believe that you will need the pendant HB donor for tetrahydroisoquinolines since it interacts with the (neutral) nitrogen in the ring to stabilize what would otherwise be a relatively unstable orientation of the pendant amide (the sulfonyl linker ‘functions’ in a similar manner). My view is that going from tetrahydroisoquinoline to the dihydroisoquinolone creates additional opportunities thereby increasing room for manoeuvre. First, you no longer need the pendant group to suppress protonation of the ring nitrogen. Second, you no longer need the HB donor for orientation of the pendant group. As a point of clarification, I must stress that these suggestions for pendant group variations only apply to the dihydroisoquinolone scaffold (I would not expect them to work for to the tetrahydroisoquinoline scaffold.
I don’t think that the relatively low potency of RAL-THA-5e0a6f1a-1 is strong evidence that the pendant HB donor is necessary when using the dihydroisoquinolone scaffold because there doesn’t appear to be sufficient space for the second methyl group even though the amide NH doesn’t donate an HB to the protein. I think that PET-UNK-c7ac4d9e-1 (easily synthesized methyl ester analog of EDJ-MED-015fb6b4-2 ) would be a better test for whether the pendant HB donor is indeed necessary since the ester and amide are isosteric.
I believe that we should be thinking of amide replacements as a means to boost potency as well as to address amide hydrolysis and see that imidazoles such as EDJ-MED-41558c53-5 have been submitted. Tautomerism can cause difficulties when trying to design an azole ring with an HB donor. I’d expect EDJ-MED-41558c53-5 to be half protonated under physiological conditions which means that only 25% of the compound is likely to be present as the desired neutral tautomer.
If you’re open to suggestions for heterocyclic amide replacements, there are some in the PET-UNK-37251634 and PET-UNK-d62a9a75 submissions and my first choice would be the 1,3,4-oxadiazole PET-UNK-37251634-3 (strictly an ester mimic although this is only relevant if the amide NH is necessary). The isoxazoles PET-UNK-37251634-1 and PET-UNK-37251634-2 mimic the amide NH HB donor with aromatic CH.
I had a look at some spiro tetrahydoisoquinolines in the CDD database and it looks like the spiro fusion doesn’t seem to adversely affect potency and hopefully the dihydroquinolone scaffold will show the same trend. Nevertheless, I would still recommend synthesizing PET-UNK-5ecb6237-1 to assess the effect on potency of substituting the central amide with cyano with a view to generating options (I’m guessing that it’ll be more difficult to engineer potency gains for the cyclic sulfones/sulfonamides than for tetrahydroisoquinolines or dihydroisoquinolones).