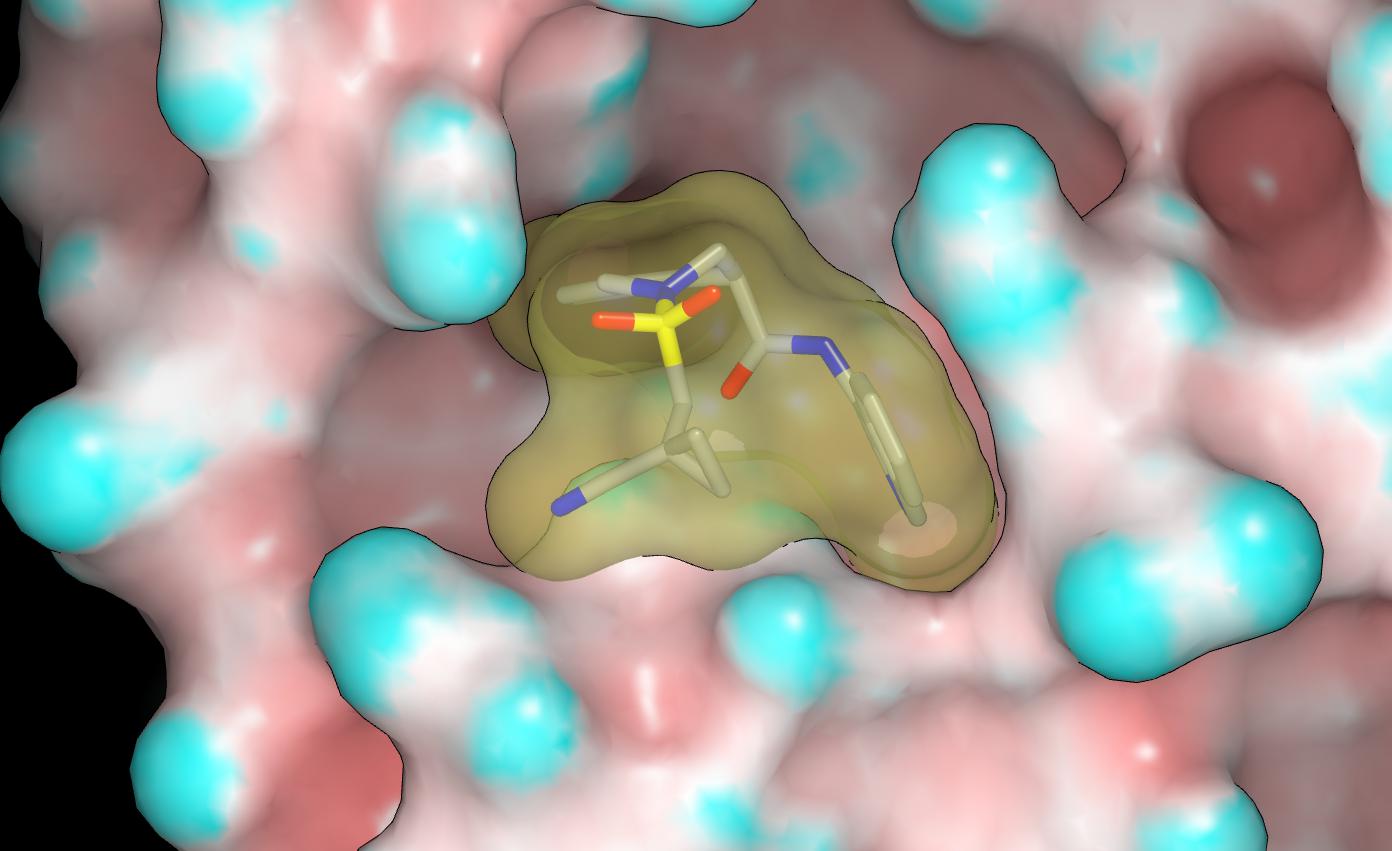
The P1090 crystal structure for the MPro complex with MAT-POS-4223bc15-23 is very interesting and I’ll mention @mc-robinson @edgriffen @Ben_DNDi @JSPEN @RGlen @frankvondelft @Daren_Fearon in case my comments are of interest. The pendant amide of MAT-POS-4223bc15-23 does not appear to make contact with the protein although there is some contact between it and the P1 isoquinoline. This suggests that the potency advantages conferred by the pendant amide result from stabilization of the bound conformation. Here’s a graphic showing the molecular surfaces of the protein (colored by curvature) and MAT-POS-4223bc15-23 (note lack of void between pendant amide and isoquinoline).
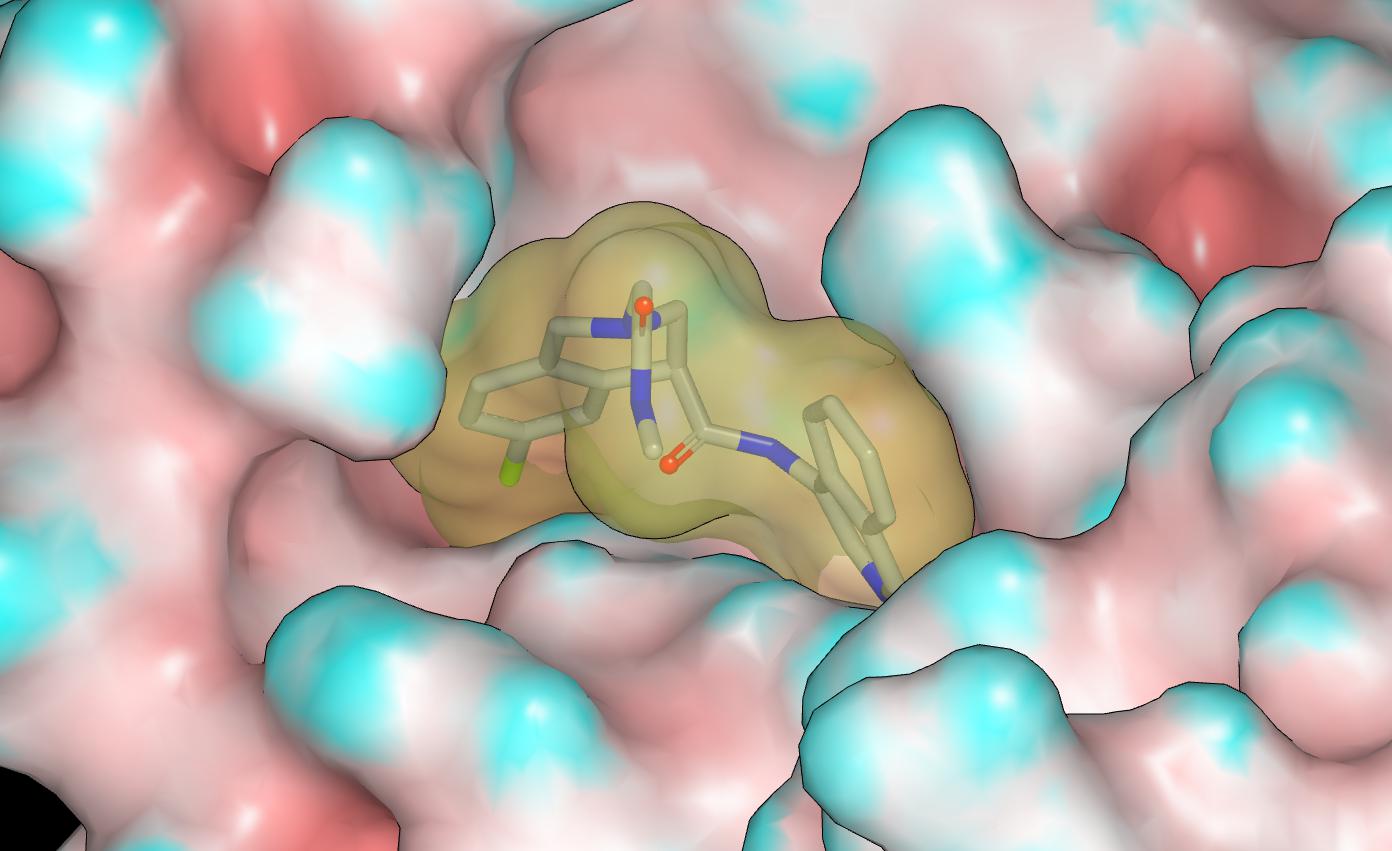
The NH of the pendant amide eclipses the lone pair of the tetrahydroisoquinoline nitrogen and this is what one would expect from looking at CSD although I’d not regard it as a genuine hydrogen bond. The NH of the pendant amide also appears to interact with the carbonyl oxygen of the other amide and, while I’d not regard this interaction as a genuine hydrogen bond, it could also be helping to stabilize the bound conformation. Given the contact between the pendant amide and P1 isoquinoline, one should anticipate a degree of non-additivity in the isoquinoline substituent SAR (especially for larger substituents) relative to the 3-chlorophenylacetamide structural prototype.
The dihydroisoquinolone EDJ-MED-015fb6b4-2 looks particularly interesting given that it is equipotent to MAT-POS-4223bc15-23. Oxa-substitution of of the benzylic methylene of the tetrahydroisoquinoline MAT-POS-4223bc15-23 removes a potential metabolic liability and the electron-withdrawing nature of the carbonyl group would be expected to protect the aromatic ring from metabolism to some extent. One more subtle advantage is that the dihydroisoquinolone nitrogen is inherently non-basic and that the substituent on nitrogen is not required to suppress protonation of the nitrogen (as is the case for the analogous tetrahydroisoquinoline). This means that the range of acceptable nitrogen substituents is less restricted for dihydroisoquinolone than for tetrahydroisoquinolone.
Some (including me) would consider an acyclic amide group, especially if primary or secondary, to be a potential liability in lead optimization. Given that the pendant amide of MAT-POS-4223bc15-23 doesn’t really function as an amide (i.e., form hydrogen bonds with protein), the design team may wish to consider amide replacements when optimizing EDJ-MED-015fb6b4-2. I suggest using a 5-membered heteroaromatic ring (see PET-UNK-37251634 and PET-UNK-f360ae44 submissions) and my specific preference is 1,3,4-oxadiazole (this article discusses why it’s preferred to the 1,2,4-oxadiazole isomer). I would not recommend using heterocycles with hydrogen bond donors (pyrazole, imidazole, triazole) since the dihydroisoquinolone scaffold doesn’t ‘need’ the hydrogen bond donor in the substituent in the same way that the tetrahydroisoquinoline does.