The recent article by Jorgensen et al may be of interest to members of the COVID Moonshot community and I’ll share some thoughts, mentioning @alphalee @frankvondelft @JohnChodera @londonir @edgriffen @mc-robinson @RGlen @JSPEN @miko_a @Daren_Fearon
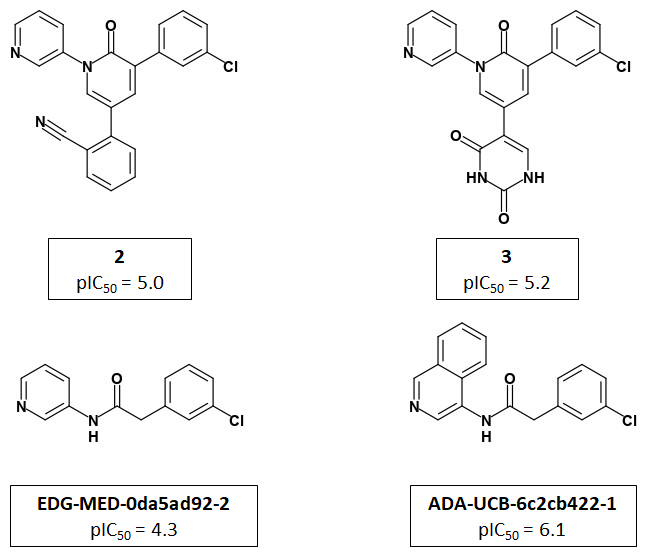
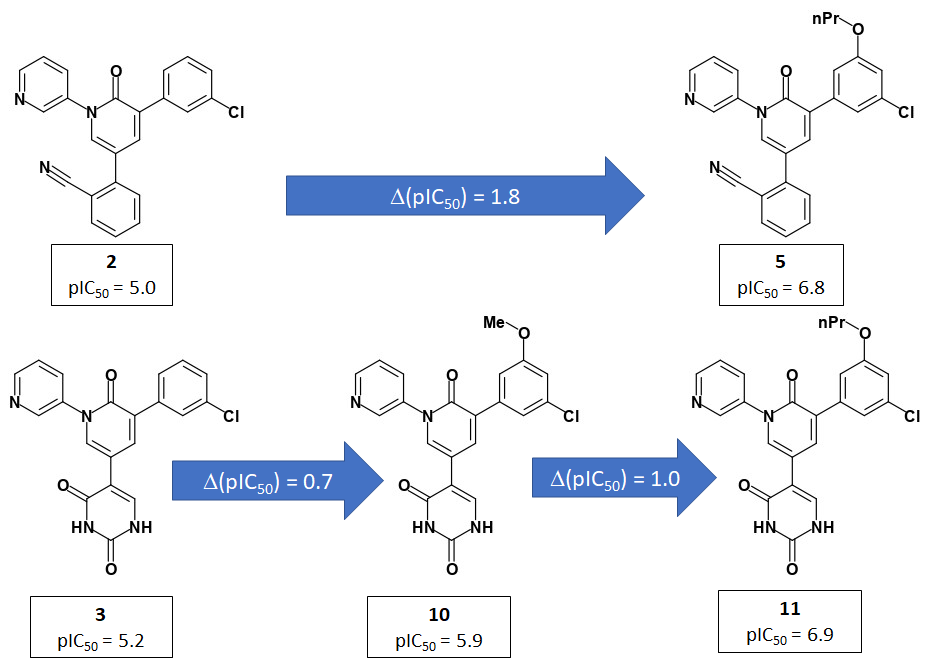
Jorgensen et al use a 2-pyridone template to present the P1 and P2 groups to the target in an analogous manner to PET-UNK-c9c1e0d8-3 which uses a delta lactam template. PET-UNK-c9c1e0d8-3 was designed as a scaffold to address the inversion of the trans/cis geometrical preference of inhibitors such as ADA-UCB-6c2cb422-1 caused by amide N-alkylation (compare with MAT-POS-bb423b95-7) with the primary objective of providing better access to the catalytic cysteine. If not attempting to target the catalytic cysteine, I would recommend using the dihydrouracil template (e.g. PET-UNK-abc197b8-1, previously submitted as the racemate VLA-UCB-1dbca3b4-18) which would present an additional hydrogen bond acceptor to the target. As discussed previously, the dihydrouracil template enables us to merge/hybridize the 3-aminopyridine-like and quinolone series. The successful use of the planar pyridone template provides support for using uracil (e.g. PET-UNK-e8933450-1), as an alternative to dihydrouracil, as the template and this would eliminate the chiral center.
Jorgensen et al use pyridine rather than isoquinoline as the P1 substituent and I would advise against automatically assuming that P1-isoquinoline will always confer a potency advantage over P1-pyridine. For example, in the Ugi series MAT-POS-f2460aef-1 (single enantiomer) and ALP-POS-02c6a514-44 (racemic) with pyridine at P1 are both more potent than the close analog ALP-POS-02c6a514-32 (racemic) with isoquinoline at P1. I see some of the benefits of isoquinoline relative to pyridine in 3-aminopyridine series as conformational and would anticipate that these benefits will be diminished when a pyridone, dihydrouracil or uracil template is used. I believe that the uracil P1’ substituent used by Jorgensen et al could readily be positioned in a similar manner using the dihydrouracil or uracil templates (but rather less readily using the chromane or related scaffolds). That said, I would want to see assay results for PET-UNK-abc197b8-1 and PET-UNK-e8933450-1 before attempting to link the P1’ uracil substituent to any of our scaffolds.
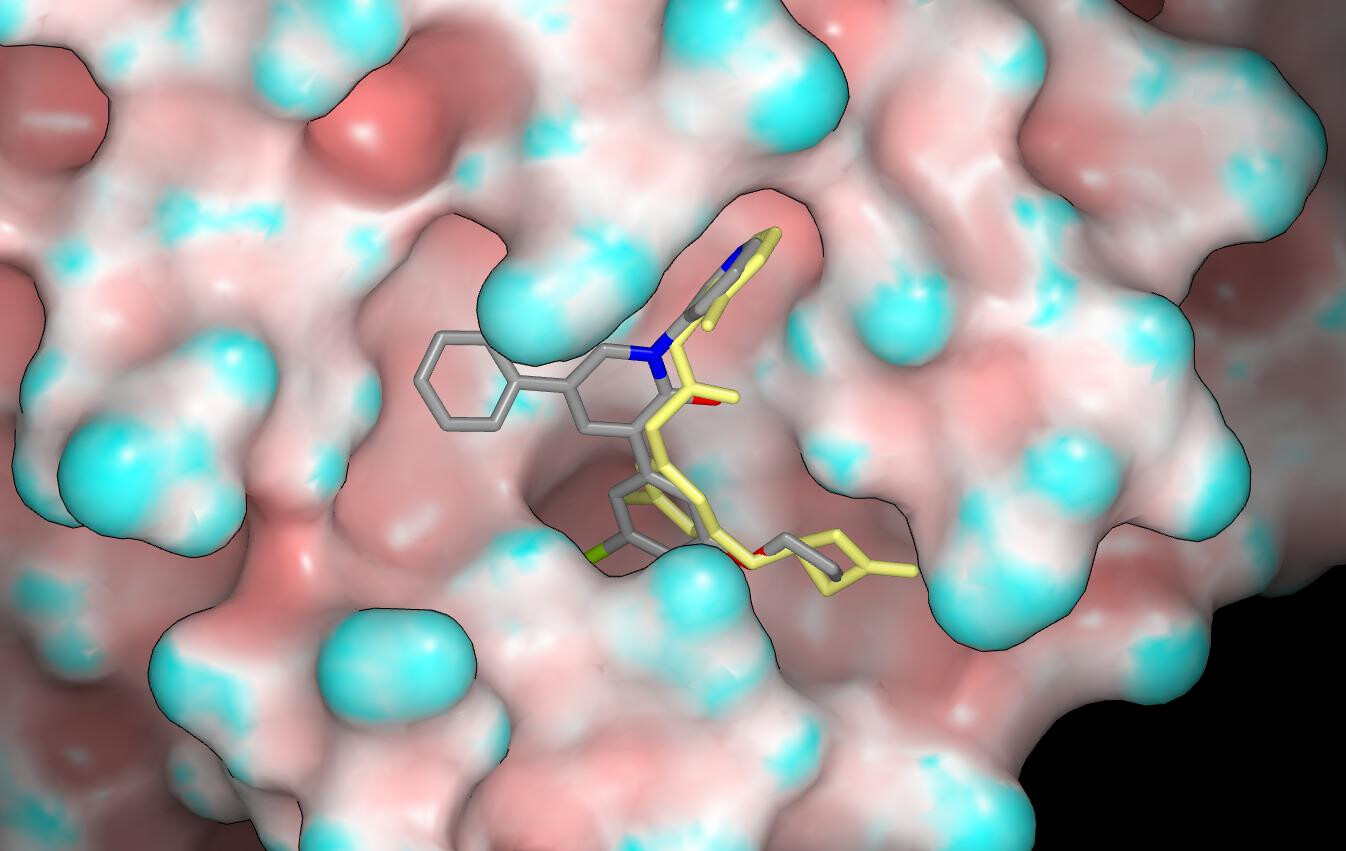
Jorgensen et al didn’t share any non-efficacy data such as aqueous solubility, plasma protein binding and metabolic stability although the activity in cell-based assays can be taken to indicate a degree of permeability. One feature of a number of the compounds that would worry me (especially for an oral agent) is the contiguous, non-polar region of molecular surface (i.e. lipophilic blob) that is associated with the P2 substituent and its pendant group. Aqueous solubility may be an issue for some of the compounds if the objective is to develop an IV agent. The crystal structures have not yet been released although I believe that they soon will be.