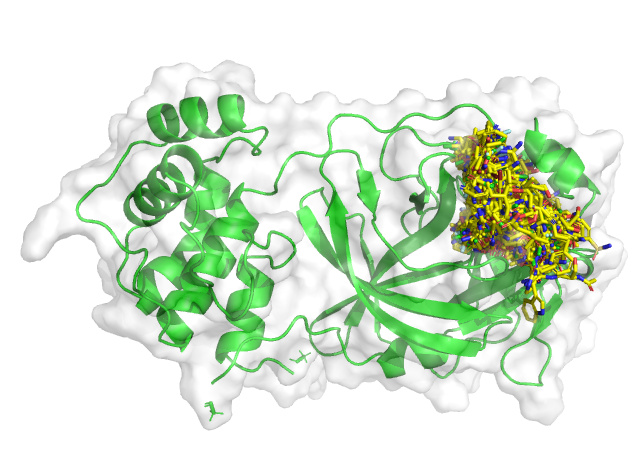
We are using the OpenEye OEDockingTK to perform ensemble docking of the updated list of all submissions to all active site and dimer interface fragment hit structures. All scripts and input/output files are in this repo:
The updated list of docked compounds can be found in this directory.
All compounds as of 18:36 PST March 31, 2020 have been redocked and scored.
This will be updated daily as new lists of submitted compounds are released.
Absolute alchemical free energy calculations for all compounds are being spun up on Folding@home right now by Matthew Hurley of the Voelz lab.
I noticed that the docking pose is quite different in some cases from the one I obtained from my workflow using glide +IFPs (SPLIF). Wouldn’t it make sense to use those poses for the AbsFEP rather than the one on github? Or would it be better/more consistent to stick to one workflow?
@bart.lenselink: Can you share your docked poses and methodology? Were you able to dock to all active site fragment structures, or did you use a specific fragment? And is there any chance you could dock the whole set (in the CSV file above) using the same methodology? If so, we could do the whole set!
Thanks for your questions: I did indeed try to redock some of the fragments but without constraints not all of them could really re-dock. Hence the use of interaction fingerprints.
For this approach however I’m not sure how useful it will be to dock the whole set, given the tailored approach: i.e. the bias that I introduce with the interaction fingerprints (SPLIF) and the single query.
One thing that I could try to setup is a similar ensemble docking workflow, using glide + shape constraints? Given that many proposed molecules are based on the fragments I could imagine this would make sense. Would this be useful?
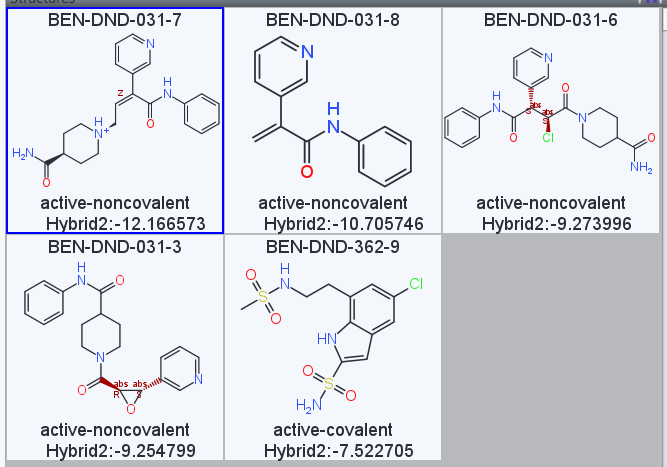
Is there a quick and simpley way to interpret this data set - i.e. is it as simple as looking at the hybrid 2 score, with most negative being “best”?
How is the “site” column assigned? Is it by hand or is it via the outcome of the docking? Just asking since some of the compounds I designed which contain covalent warheads have been assigned as “acitve non-covalent” site, and vice versa (i.e. componuds with no warhead have been asigned as “active-covalent” site…
Since you have multiple methods, and no way to validate which method is better, would you want to prioritize molecules that dock with high scores by both methods?
A more negative Hybrid2 score is better. We were mainly looking to exclude designs that clearly did not make reasonable interactions with any if the inspiration structures listed by eliminating those that scored very poorly (around -6 or greater). With compound sets this small, it’s not clear that docking scores like this are sufficient to really identify very potent binders, so we were also looking for diversity and other criteria at the first stage.
The <fragments> SD tag in the latest docking results lists the set of user-specified inspiration fragments where the molecule scored best when fit into the co-crystal protein structure for that fragment. The site is then the classification of the fragment—whether the fragment was a noncovalent active site binder, a covalent active site binder, or a dimerization site binder.
Hi Anthony,
As some examples here are 4 compounds which contain various covalent warheads designed to hit CYS145 in the active site which for docking purposes have been assigned as non-covalent, and also one example which is assigned as covalent but has no warhead.
Out of interest, when you dock are you manipulating the targets to satisfy the post-covalent-linking structure e.g. docking the epoxide compound BEN-DND-031-3 as the “sprung-open” secondary alcohol, and docking the acetamide-like compounds as fully saturated around the double bond?
Hi Ben, Thanks for this. We are currently reviewing the way we filter covalent compounds submitted. I will check that when available, the most updated spreadsheet doesn’t have the same issues.
I see that there is quite a bit of discussion already around the covalent/non-covalent docking issue amongst the ensemble docking reported here. But I would like to ask, because it isn’t clear to me, have all the compounds in this collection been submitted to non-covalent docking only? If non-covalent docking has been performed here, then were the warhead-containing submissions docked with their warheads intact?