Category for discussing potential designs based on the latest data. All discussions regarding simulations (docking, FEP, ML, etc…) should also take place here.
Which amino acid side chain residue of the protein is supposed to attack the chloro-methylene portion of the covalent inhibitor? Would try to understand better in order to replace this function (potential genotoxicity, and the therapy is supposed to least several weeks…) into a non covalent one. Thanks
Hi, I believe it is a cysteine. Scheme shown here. Please note that it is a bit hard to tell in some of the Fragalysis structures that the fragment is covalent.
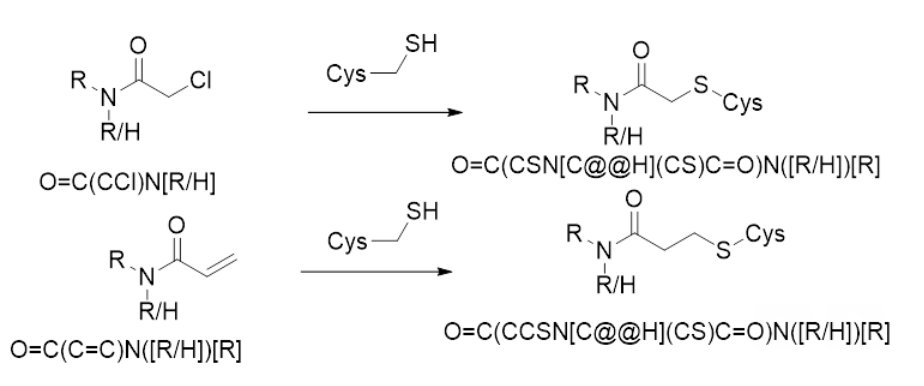
@pgz may have more comments
So just supposed mechanism of action and positive response of the fragment analysis, if I understood well. Could it mean that also non-potential alkylating fragments, but endowed with similar polarity (or other molecular descriptors) of chloroacetamide could also fit well in the supposed subpocket? Could it be a possibility in which the molecular space can be explored in the hit design?
Hello,
Matt already posted the answer to part one of your question.
Since the initial covalent crystallographic hits were fragment-sized, I’d expect them to have a very low affinity without the electrophile. This means you’d probably need to gain significant new interactions if you want to get rid of the electrophile in your new designs. Also, I’m not sure in this case if it would be worthwhile to even have a the N-acetyl motif, if it doesn’t contribute any H-bonds or other significant specific interactions in your designs. Any other thoughts on this by other members here?
You make a good point re. toxicity, however one that is frequently brought up. If you want some more background info I would suggest reading the sections on chloroacetamides and acrylamides in this review (there are others, too many to post here): https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b01153
My quick personal opinion: I think there would be possibilities to engineer out issues of high warhead reactivity later on, but I understand where the concerns are coming from.
Best
Paul
Hi all,
It is possible to have a warhead and be drug-like. Odanacatib was a Cys-protease inhibitor (Cathepsin K) with a electrophile warhead tested in Phase III clinical trials. In that case the warhead was a nitrile which is additionally protected by a cyclopropyl. The compound fell because MSD lost interest.
best Joost
So you think that, in this early phase, the main design effort would be to keep the covalent inhibition with chloroacetyl mojety (or other electrophiles) and explore the chemical space of the potential scaffolds?
I think a covalent drug would be a good idea in this case. Since we are targeting a viral protease, this destructive mechanism could work to our advantage. The covalent bond helps increase the potency and, in case of an irriversible inhibitor, pharmacodynamic drug properties.
Ofcourse it is possible to swap out the chloroacetamide for a softer electrophile if specificity/toxicity is a concern. There are also electrophiles that form a reversible covalent bond.
Some members of my group have produced a predicted, glycosylated version of the spike protein. See an image of it at the link below. If you are interested in working with them, I can put you in touch.
I tried to embed an image here, but it won’t show.
Very topical to current call, see:Second design call: focus on covalents and merging with non-covalents