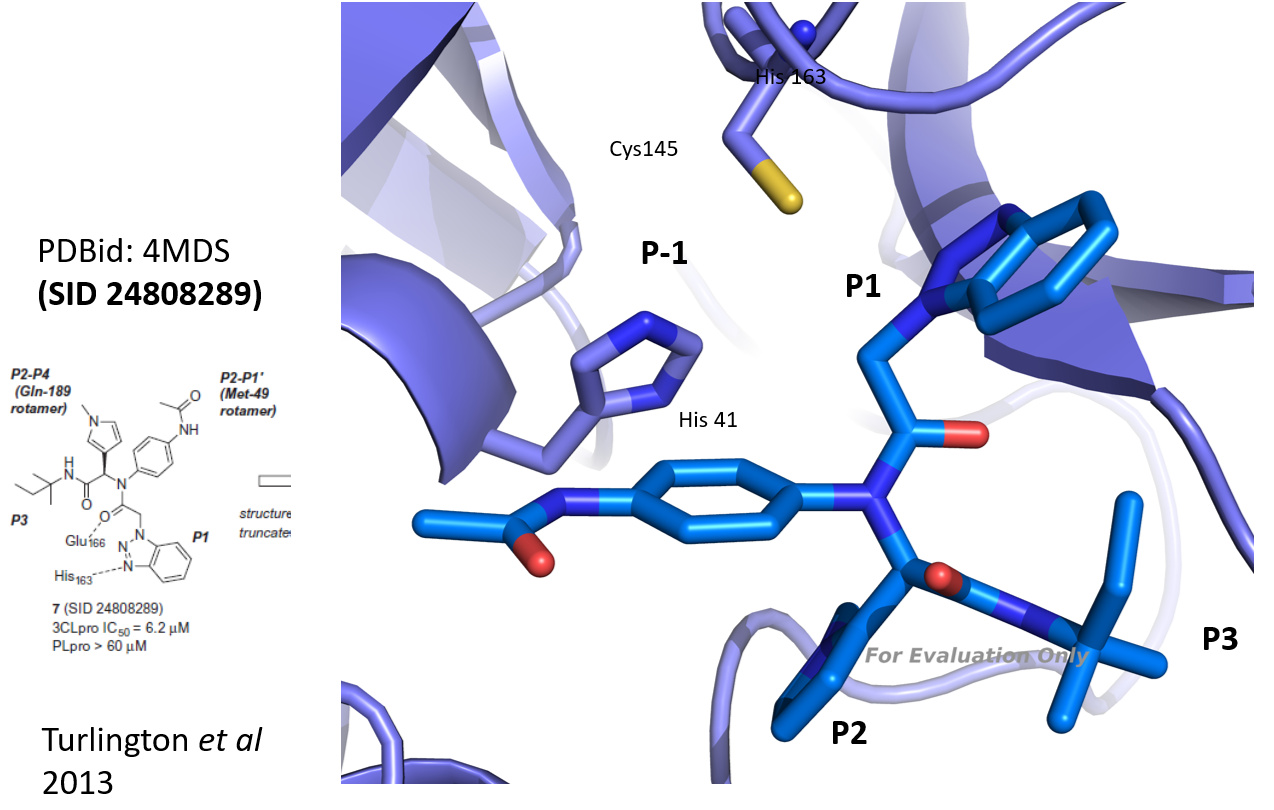
@mc-robinson I took the 3D structure of SID 24808289 (Turlington et al 2013, PDBid: 4mds, see figure) and compared it to the results of the analog search you performed. As you already pointed out, the series is quite interesting since representatives can bind with high potency even though not covalent. Based on a visual check (and a bit of imagination), I made a selection (in the attached powerpoint). I would keep the benzotriazole (which binds to Cys 145), and the amide and tertiary amine and then bring in diversity in the hope to find a molecule that addresses the P-1, P2 or P3 pockets efficiently. Hope this helps. Let me know - best Joost
existing_inhibitors_Enamine_selection_JU.pdf (455.3 KB)