I think the activity across the different SARS proteases is very encouraging since we might anticipate that other SARS proteases will need to be inhibited in the future.
Here is some more SAR that might be of interest:
Dipeptide aldehydes tend to be significantly more potent that the corresponding dipeptide nitriles. Azadipeptide nitriles are very potent when compared to the corresponding dipeptide nitriles (I think this because the adducts formed from the azadipeptide nitriles are more basic although I wouldn’t claim to fully understand this). Here is an article from a group (Nequimed) in Brazil that I work with:
@pwkenny Very interesting. The 11a/11b from the Dai et al paper look like the ‘Pfizer-patent inhibitors’ above. They have nice potency with their 40-50 nM IC50 (although this doesn’t count for covalents, I know). It would be interesting to replace the aldehyde with a nitrile (or aza-nitrile), as mentioned in the Silva et al warhead paper you quote. Guessing from the x-ray structures, you would have to replace C=O of the aldehyde directly with C=N. If potency is good, the next step would be to make it less peptidomimetic. Actually you would be in the same starting point as the inventors of odanacatib when they started, as in the paper below (and that drug made it to Phase III)
Converting a peptidomimetic nitrile-based CatK inhibitor into a Phase III drug (odanacatib), https://pubs.acs.org/doi/abs/10.1021/jm0301078
(guess I’m getting off topic here -sorry for that)
Wow @JoostU and @pwkenny, this is amazing. This should become a review article!
Thanks especially @JoostU for making my info much more presentable, and for providing some nice PDB views.
I have some more reading to do based on your suggestions, but I think one question to put to you two and the crowd is what molecules of these do you think should be immediately made and re-tested?
We have also considered making many “matched pairs” of warheads on each scaffold to test the warheads. And additionally, we would likely look to get these inhibitors into crystal structures.
Just wanted to flag up a couple of points. If a covalent inhibitor binds reversibly then Kd can be measured and IC50 and Ki can be interpreted in the convential manner. It is even possible to use ITC to study the binding of covalent inhibitors that bind reversibly.
The hydrogen bond donor is not eliminated in odanacatib but the amide in question is replaced with an amine that is of low basicity on account of the CF3 group. However, the hydrogen bond donor of that amide can be eliminated by using the cyclohexane dicarboxamide scaffold:
Thanks Peter for pointing that out. I was too quick with my remark about Kd. I just posted the amide-aryl replacement picture as it was a first step in something that was quite a long journey by Merck Frosst to generate odanacatib from a peptidic inhibitor. In my time at MSD, I got to work on the project a few years and was really impressed. Quite a waste that such a good inhibitor got archived. Merck Frosst also had to solve quite some issues that only popped later in more advanced biological models. I think there is a lot to learn from this drug discovery trajectory for the Covid project. Here are a few more links, which I’m sure you are aware of https://pubs.acs.org/doi/abs/10.1021/jm800610j https://pubs.acs.org/doi/abs/10.1021/jm058198r
There certainly is a diversity of scaffolds, even when looking just at cathepsin K, and there could be value in seeing how some of the different scaffolds might fit into the SARS-Cov-2 protease. Pharma/biotech companies, who have been making noises about wanting to help, have large amounts of proprietary data in their databases and samples in their corporate collections.
@mc-robinson: I’ve docked this list using the OpenEye OEDocking noncovalent ensemble hybrid docking, using the bound fragments to pose these inhibitors in all fragment structures. All data is here:
The SDF file that @Waztom and @reskyner may be interested in is downloadable here. This isn’t yet in the new format since I think the SDF fields haven’t yet converged on specific tags and specifiers yet, but I’m happy to convert to a more convenient format when needed!
I finally got around to taking a look at past SARS inhibitor compounds that are not directly purchasable – to see if anything was in Enamine REAL or WuXi Galaxi close to the old compounds (Thanks to @Franca for the BioSolveIT license!) . I’ve collected that data in the large pdf here (sorry for the weird format, originally the document was a lot of code with plots mixed in). Do let me know if you see anything worth ordering. I have circled compounds I think may be worth ordering, but that was a very quick read over right before I got back to coding – and I would appreciate your input.
@JohnChodera also pointed out that the anilide inhibitors in this paper (see graphical abstract) have many similar compounds in REAL (including the compound below itself).
Some look quite lipophilic and maybe PAINs (Cl-nitroaryl group) but possibly worth a punt if a few can be readily synthesised. I notice, in the Nature paper, detergent assays were used to rule out aggregation effects. If these are purchased, is it worth rerunning to rule out aggregation effects?
(I started writing this a week ago, but forgot to come back to the browser tab + finish it!)
The 3CL-protease is well conserved between different viruses. Most of the active drug-like molecules are directly derived from the natural substrates. The best work I found on looking at QM/MM interactions with the substrate are the work of Paasche, who spent a PhD with Bernd Engels looking at SARS-CoV(-1).
The paper is probably the most dense high-level collection of their work.
Paasche, A., Zipper, A., Schäfer, S., Ziebuhr, J., Schirmeister, T., Engels, B. (2014). Evidence for Substrate Binding-Induced Zwitterion Formation in the Catalytic Cys-His Dyad of the SARS-CoV Main Protease. Biochemistry, 53(37), 5930–5946. https://doi.org/10.1021/bi400604t
But the details are better explained in his (freely available as a PDF below) thesis:
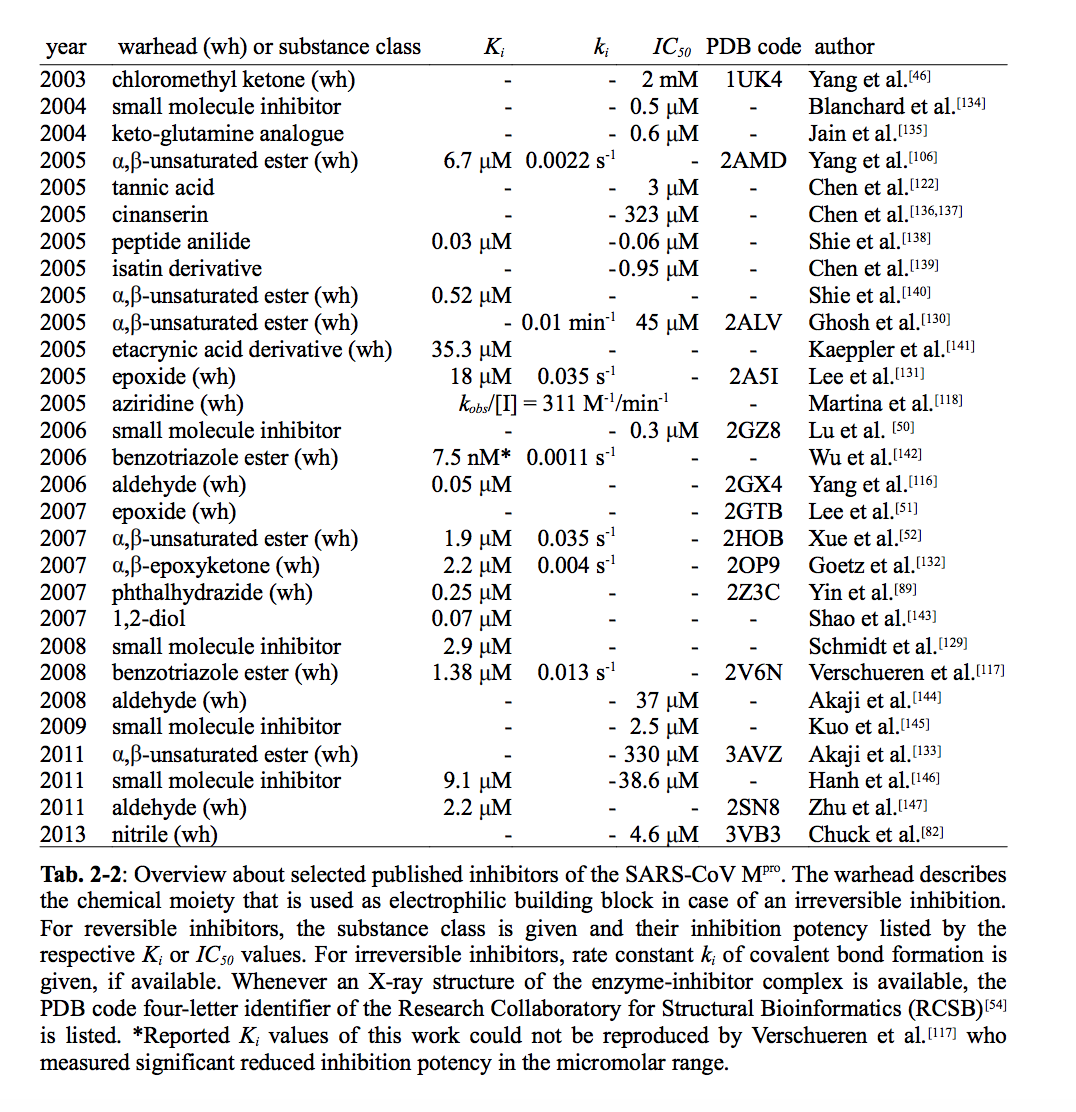
The thesis is probably also a good introduction to covalent inhibitors for this target. (For instance, see table 2-2, which lists all inhibitors as of 2013, with reference, PDB code, and activity.)
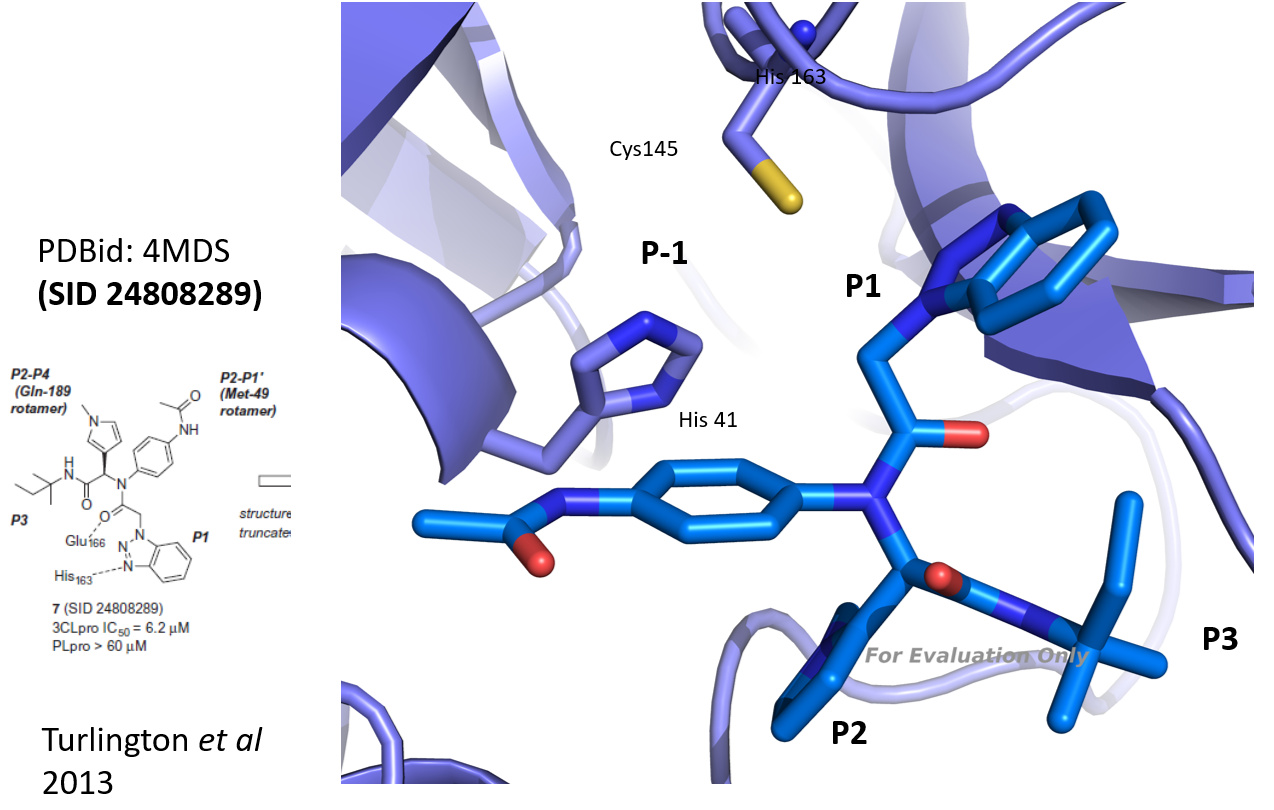
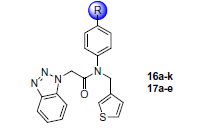
@mc-robinson I took the 3D structure of SID 24808289 (Turlington et al 2013, PDBid: 4mds, see figure) and compared it to the results of the analog search you performed. As you already pointed out, the series is quite interesting since representatives can bind with high potency even though not covalent. Based on a visual check (and a bit of imagination), I made a selection (in the attached powerpoint). I would keep the benzotriazole (which binds to Cys 145), and the amide and tertiary amine and then bring in diversity in the hope to find a molecule that addresses the P-1, P2 or P3 pockets efficiently. Hope this helps. Let me know - best Joost
Benzotriazole has a pKa of 8, is known to be activated via general acid catalysis and well positioned to react with CYS145. Can you really exclude formation of a covalent linkage with CYS145? Did the authors do experiments to exclude this hypothesis (apart from this XRD structure)?
Hi all,
In trying to answer the question above, I came across two reviews by Pillaiyar et al., who weren’t mentioned yet as far as I know
Great and extensive review on SARS protease targeting: https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.5b01461
And a very recent, more broad overview by the same author
In the last work I encountered AG-7088/ rupintrivir, a Pfizer drug that was already tested against the SARS protease in 2003. It made it as far as Phase II.
@JHullaert. Nice remark. I reread Turlington et al. and they did not do further experiments to look at reactivity with Cys 145. Also found the work below, presenting new benzotriazole series in the wake of the Turlington work. But IC50s are disappointing there (> micromolar). Actually only Turlington’s compound 17a (50 nM) has any decent potency. If it was a general reaction between benzotriazoles and Cys 145, I would expect a bit better IC50s overall. Nevertheless, 17a remains interesting, I think. An X-ray structure would help.
(Of course the benzotriazole series we talk about have a different binding mode than the suicide inhibitors with benzotriazole as a leaving group that are also mentioned above, or could 17a have a suicide inhibitor-like mechanism?)
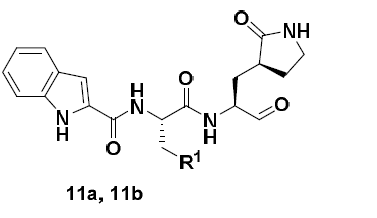
Somebody may have already flagged these up but, just in case not, there are a couple of aldehydes (IC50 values of 40 nM and 50 nM) and there are crystal structures although these don’t seem to have been deposited yet: https://doi.org/10.1101/2020.03.25.996348
The structure shown with the 30 nM Ki could be functioning as acylating agent (electron-poor aromatic ring facilitating nucleophilic attack on anilide cabronyl) and it’d be good to get crystal structure so that we can get a better idea of binding model. The chlorine looks like a good halogen bond donor. If this is the case then [Cl->Br] would be expected to lead to an increase in affinity. Oxidation of the catalytic cysteine thiol is always a concern when studying non-covalent binding to cysteine proteases).