Topic automatically created for discussing the designs at:
https://covid.postera.ai/covid/submissions/PET-UNK-a17c93d1
This design aims to improve on the @edgriffen design EDJ-MED-c314995a-1 (fluorescence pIC50 = 6.6) which is likely to bind in a relatively high energy conformation (by analogy with the JOR-UNI-2fc98d0b-12 X10236 crystal structure) and I’ll also mention @mc-robinson @frankvondelft @JohnChodera and @miko_a (who has just submitted the MIC-UNK-cdc2493e design).
Here are a couple of torsion distributions (absolute values used) from analysis of the Cambridge Structural Database (CSD):
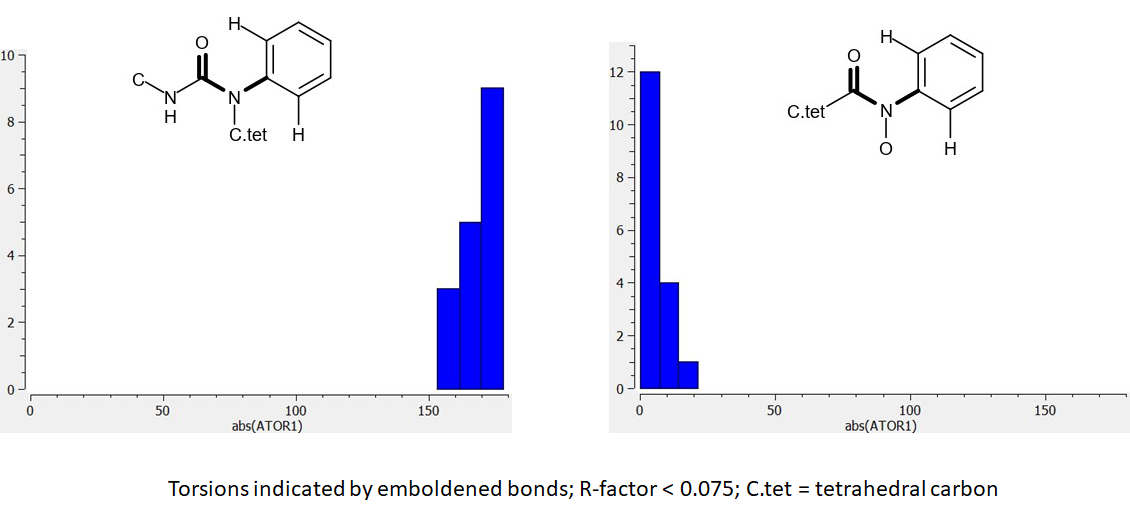
There were insufficient data in the CSD for urea analogs with oxygen bonded to nitrogen and I’ve used the amide equivalent instead. The abs(TOR1) value corresponding to the JOR-UNI-2fc98d0b-12 urea ligand in the X10236 crystal structure is 13.5 degrees. This observation indicates that the ligand is likely to be binding in a relatively high energy conformation. Comparison of the two distributions shows how the bound conformation can be stabilized by substitution of oxygen for methylene.
I generally consider the binding of a ligand in a relatively high energy conformation as an opportunity (this article shows how we used analogous information in the AZ PTP1B project). The large difference in potency between MAT-POS-bb423b95-7 (fluorescence pIC50 = 4.5) and ADA-UCB-6c2cb422-1 (fluorescence pIC50 = 6.1) is likely to be due to the change in the cis/trans geometric preference of the amide that results from N-methylation.
1 Like
Pete, do you have any other options for that torsion that avoid the N-O bond - I have the traditional medicinal chemists fear of idiosyncratic hepatotoxicity.
1 Like
H Ed,
One very obvious option would be to attach another aromatic ring to the amide nitrogen and this would effectively hybridize the series with the ‘benzotriazole’ series. I’d actually been thinking about this although I wanted to see how what the conformations for the Ar2NC(=O) substructure might look before submitting a design.
Generally, I’d be wary of N-O single bonds like these although they do occur in approved drugs (hydroxyurea, belinostat, panobinostat, selumetinib, binimetinib) according to drugbank. The 1,4-substitution of one of the aromatic rings in typical representatives of the ‘benzotriazole’ series would worry some because of the possibility of forming ‘quinoid’ metabolites a-la-paracetamol.
From a more general safety perspective, achieving efficacy with a low dose is likely to make some problems go away (particularly for covalent inhibitors). If you are planning for health care workers to take a drug prophylactically then it will have to be squeaky-clean. Given that aromatic nitrogen at the periphery of the molecular structure is a pharmacophore element that is common to a number of the series under consideration, it would be prudent to assess CYP inhibition at this stage of the project. It would also be prudent to consider safety issues connected with hydrolysis products (e.g 3-aminoisoquinoline).
1 Like
Thanks for the thoughts @pwkenny, possibility of CYP issues has definitely crossed our minds given that potency seems to be (perhaps largely) derived from strength of H bond with that aromatic N. Results of CYP assays should be here in next 1-2 weeks!
Hi Matt, I look forward to seeing the CYP results. If there are problems, we’ll hopefully be able to extract some SAR for CYP inhibition that can be used to address the issues.
Hi Ed, looks like a 1,1-diphenylurea MIC-UNK-d36ab305-5 / FRA-DIA-13af2da5-1 has been suggested by @miko_a and @frankvondelft . Having two sp2 carbons bonded to nitrogen gets round the problem of an sp3 carbon on the nitrogen EDJ-MED-c314995a-1 of having the stronger preference for being syn to carbonyl oxygen. There are potential issues around the relative orientations of the two phenyl rings and I can investigate using the CSD if people are interested.
I’m afraid the point is moot to some degree. I expect that additional cycle like in chromane system will increase potency independently of other substituents, which leaves amides of 4-aminoisoquinoline (or similiar) as the only possibility. I’ve submitted these structures mainly because these ureas and amides are easier to make and also this makes pairwise comparison with what already has been assayed possible.
When there are found good substituents filling P1’ pocket (and/or P4 pocket) using urea backbone I think next step could be cyclization and synthesis of appropiate chromane or similiar compounds. In that case conformational prefereces could be at least crudely predicted using something like A values.