Topic automatically created for discussing the designs at:
https://covid.postera.ai/covid/submissions/PET-UNK-1901c25b
I’d like to provide some more detail for design 2 of this set (and also apologize for design 3 being a duplicate of a previously submitted design). This design places the methylsulfonyl carbon in a similar position to the corresponding carbon in the AAR-POS-d2a4d1df-1 fragment and I’ll mention @frankvondelft who may be interested.
I think the N-S torsion is wrong in the fragment structure (it’s analogous to the glutamine side chain issue) and here’s a pic of the modeled fragment in the crystal structure:
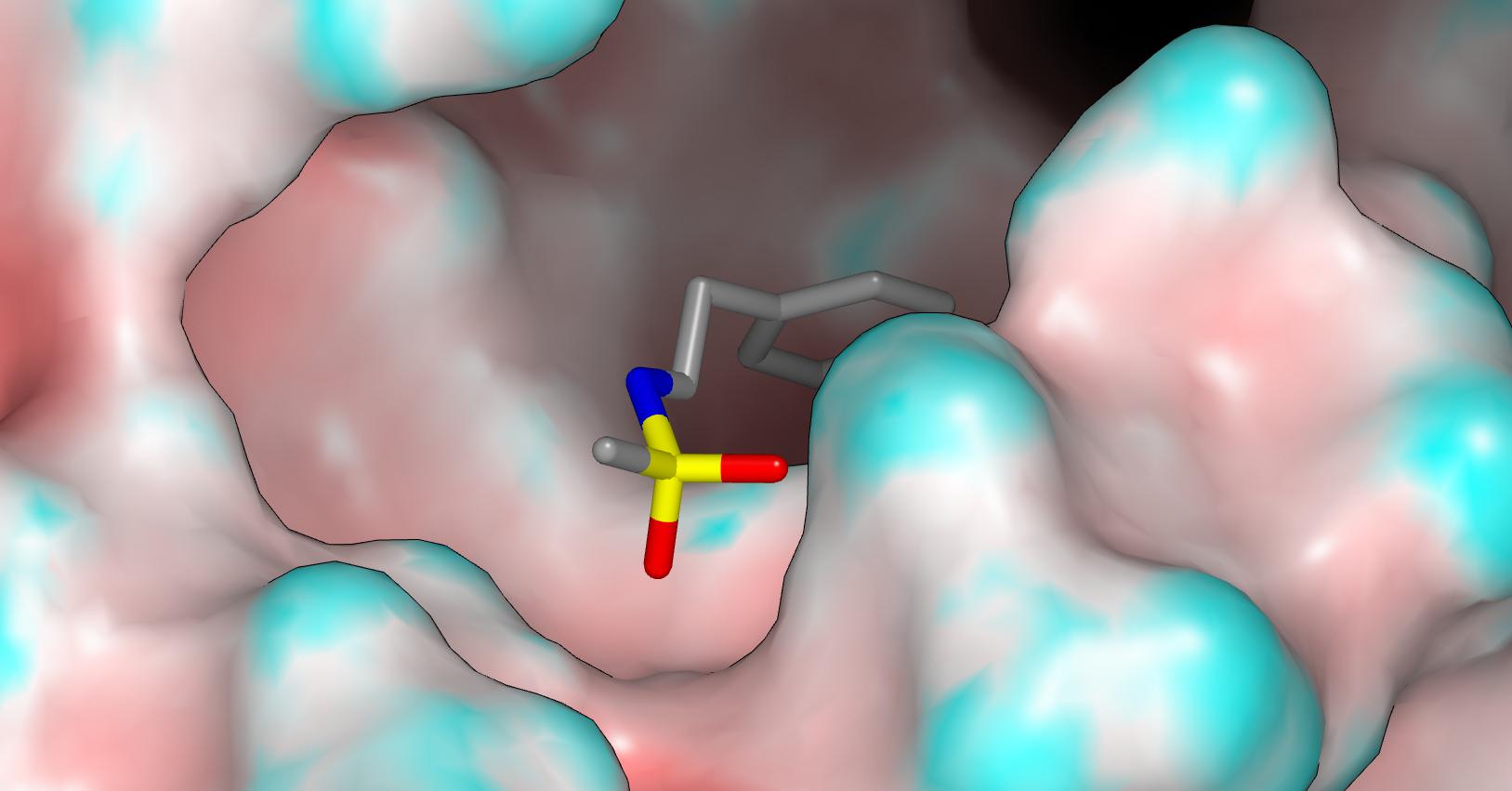
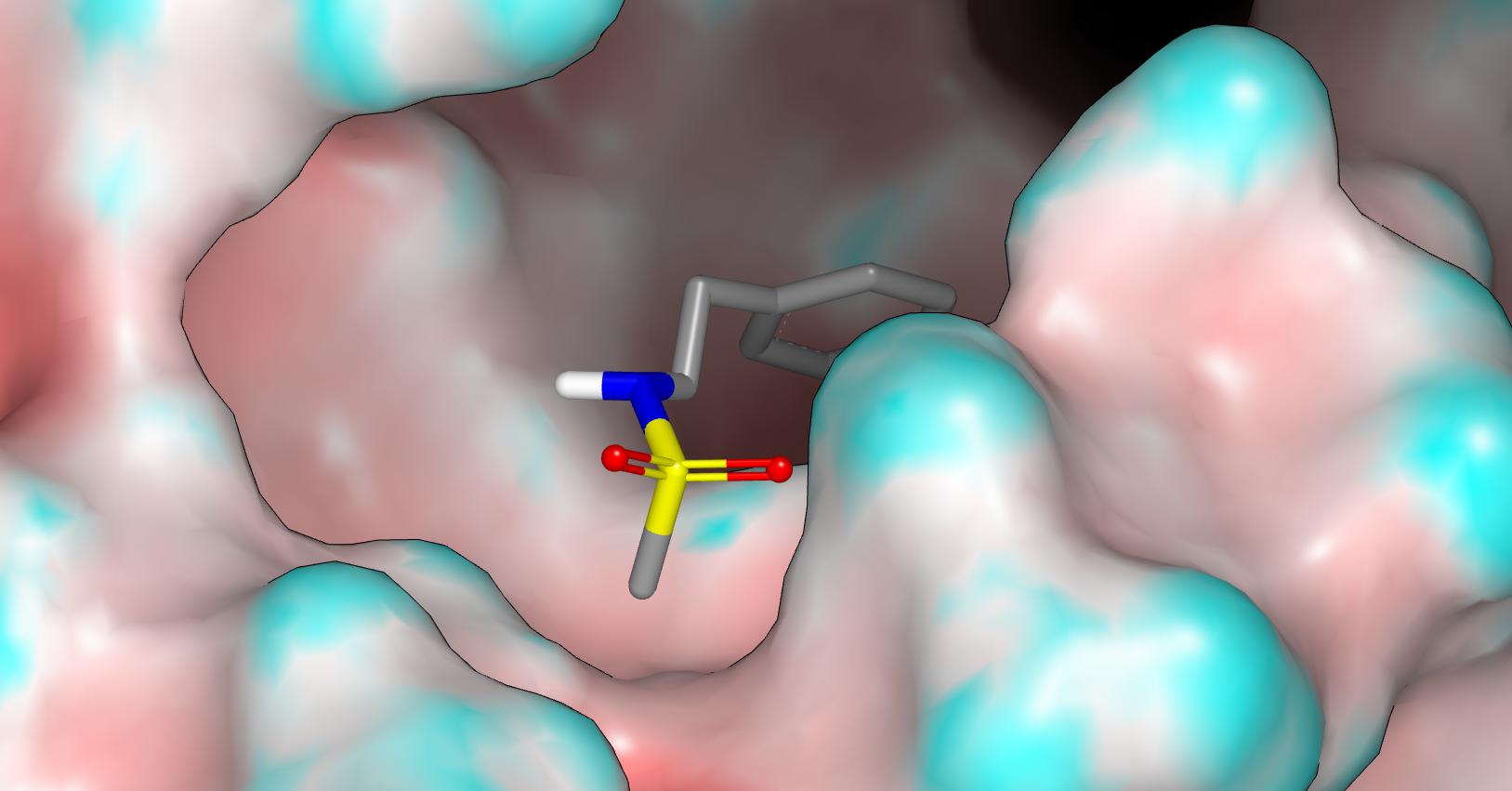
Here’s a pic of what I think is the actual conformation and this is more in line with what would be expected from sulfonamides in small molecule crystal structures. If it is of interest, I can share some torsion distributions. I should stress that I consider this fragment structure to be highly informative and it strongly influenced my decision to design the methylsufonyl group into the structure.
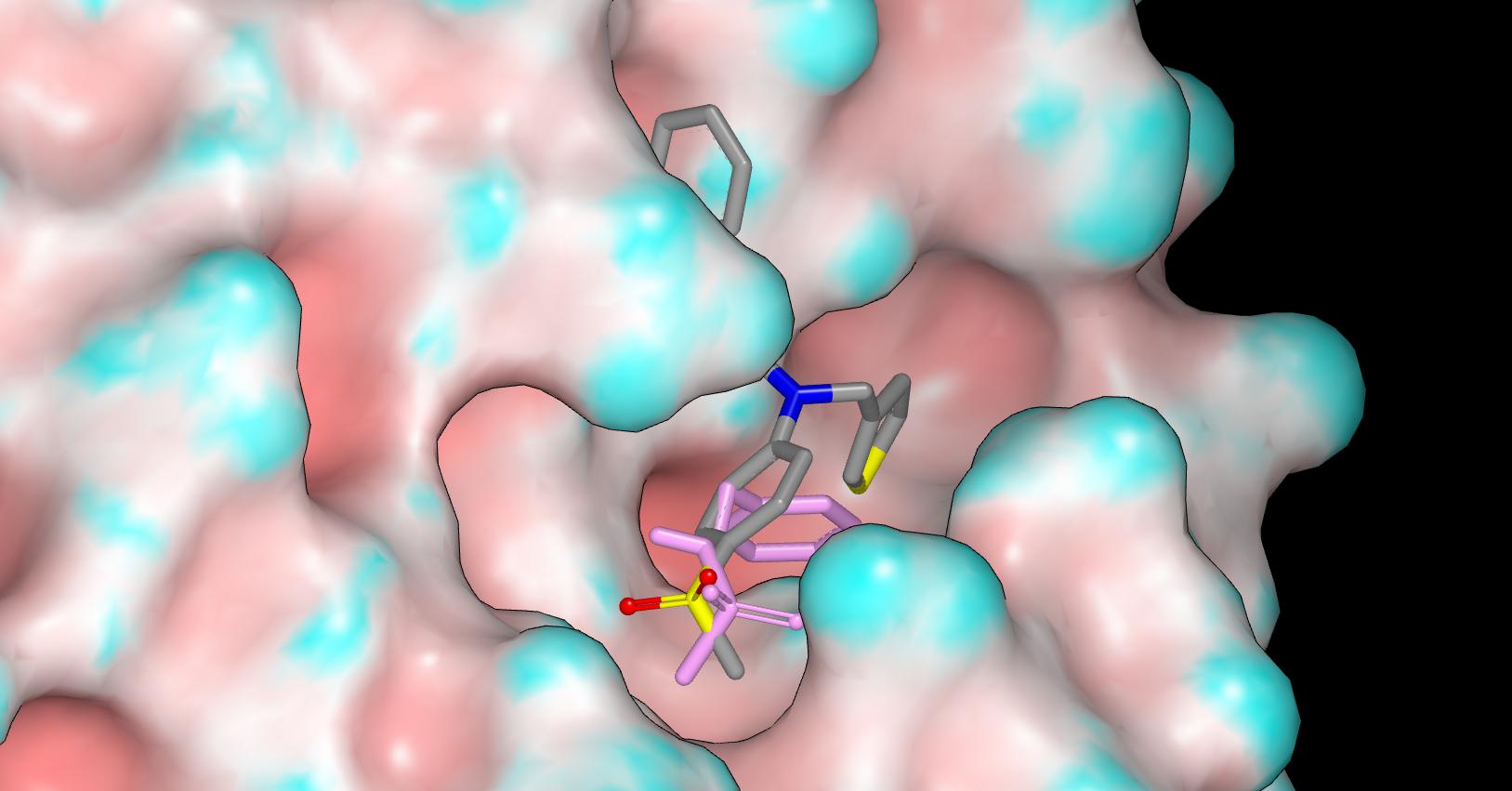
Here’s a pic of the designed structure overlaid with with the fragment in a rather lurid hue.
The methylsulfonyl group is a good substituent from the ADME perspective and this article may be of interest.
Thanks Peter - we went to quite a lot of trouble at the time to match the electron density, but obviously, it is sometimes not 100% clear. But feel free to take a look yourself, we tried to make it as easy as possible for others to view it (which doesn’t mean it’s one-click-easy, but instructions are clear).
https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Electron-density-evidence.html
Let us know if you still believe the conformation is wrong. (It’s massively and depressingly tedious to update coordinates, btw, so I won’t promise an instant fix…)
Thanks for getting back to me, Frank, and the issue is whether the electron density allows an oxygen atom to be distinguished from a methyl group. I’m not questioning the conformation in terms of ‘generic’ non-hydrogen atom positions (I’m assuming that standard bond lengths are used when fitting density). I’ll do some analysis of sulfonamide torsions from the CSD and post in the general forum early next week. I believe that the CCDC has software for assessing ligand torsions. I’m definitely not suggesting that you even consider updating coordinates at this stage.
One a semi-related note, I will attempt to dock on this compound (IC50 = 0.6 µM in the fluorescence assay but completely dead in RapidFire) and will explore the possibility that the methyl of the acetyl group overlays with (what I think is) the methyl group of the sulfonamide. I’ll keep you posted.
Yes, I’m also betting that the piperazine is sticking into P1’ rather than P2. But it’s in crystals now, so we’ll hopefully soon know.
Excellent and good luck getting a great structure!