Topic automatically created for discussing the designs at:
https://covid.postera.ai/covid/submissions/FRA-DIA-3f9ae552
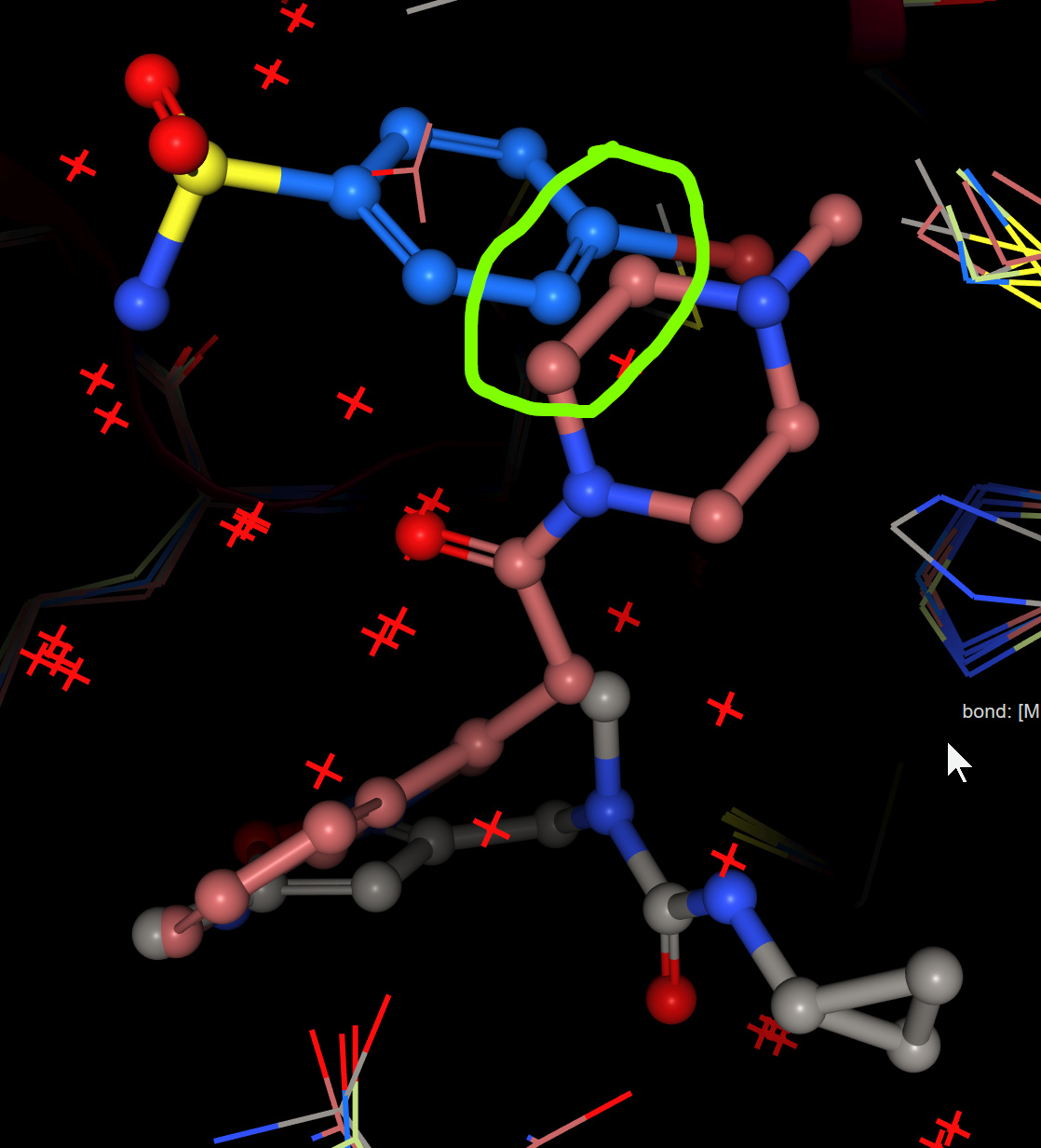
Compound 1, fusing x1093 (piperazine) and x0946 benzene:
https://fragalysis.diamond.ac.uk/viewer/react/projects/35/73
Screen grab:
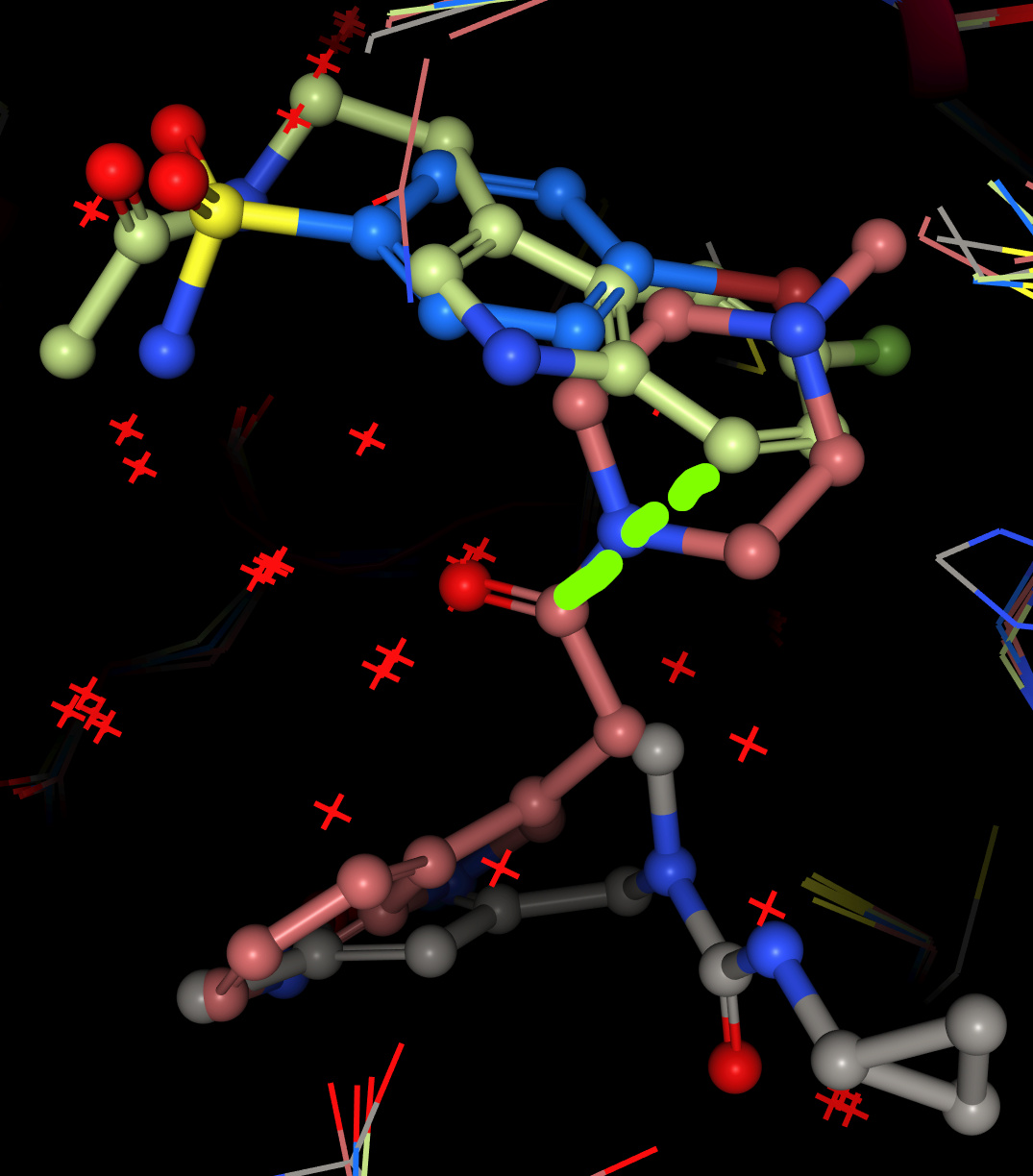
Compound 2, replacing that piperazine/benzene merge with the indole of melatonin-like x0104, but attaching the sulfonamide of x0946:
https://fragalysis.diamond.ac.uk/viewer/react/projects/35/74
Screen grab - the indole would need to rotate to accommodate the indicated connectionm, which is quite long for the fragments as they appear in the crystals:
Preamble: I cheated by being literate
So, the by far the biggest problem with Fragmenstein is “over-inspiration” (too many diffuse inspirations) and “off-inspiration” which is a problem fairly unique to the covid project, wherein the an inspiration is not an inspiration, but was for another compound or the wrong ID was selected. In normal mode (partial merge to avoid over/off-inspiration) a few too many fragments get refused. As I assume that you mostly care about stability not how picky is the algorithm, I am cheating and reading what you wrote and selectively feeding the compounds are running in unmerging mode (where each possible fragment combination is mapped like a suduku puzzle) as its the safest. I actually have been using this set as test bed for the last 3 days actually and made me realise a few things.
—so thanks!
FRA-DIA-3f9ae552-1
For FRA-DIA-3f9ae552-1, against x0397, x0946, x1093 (no x0104), the result is a happy molecule
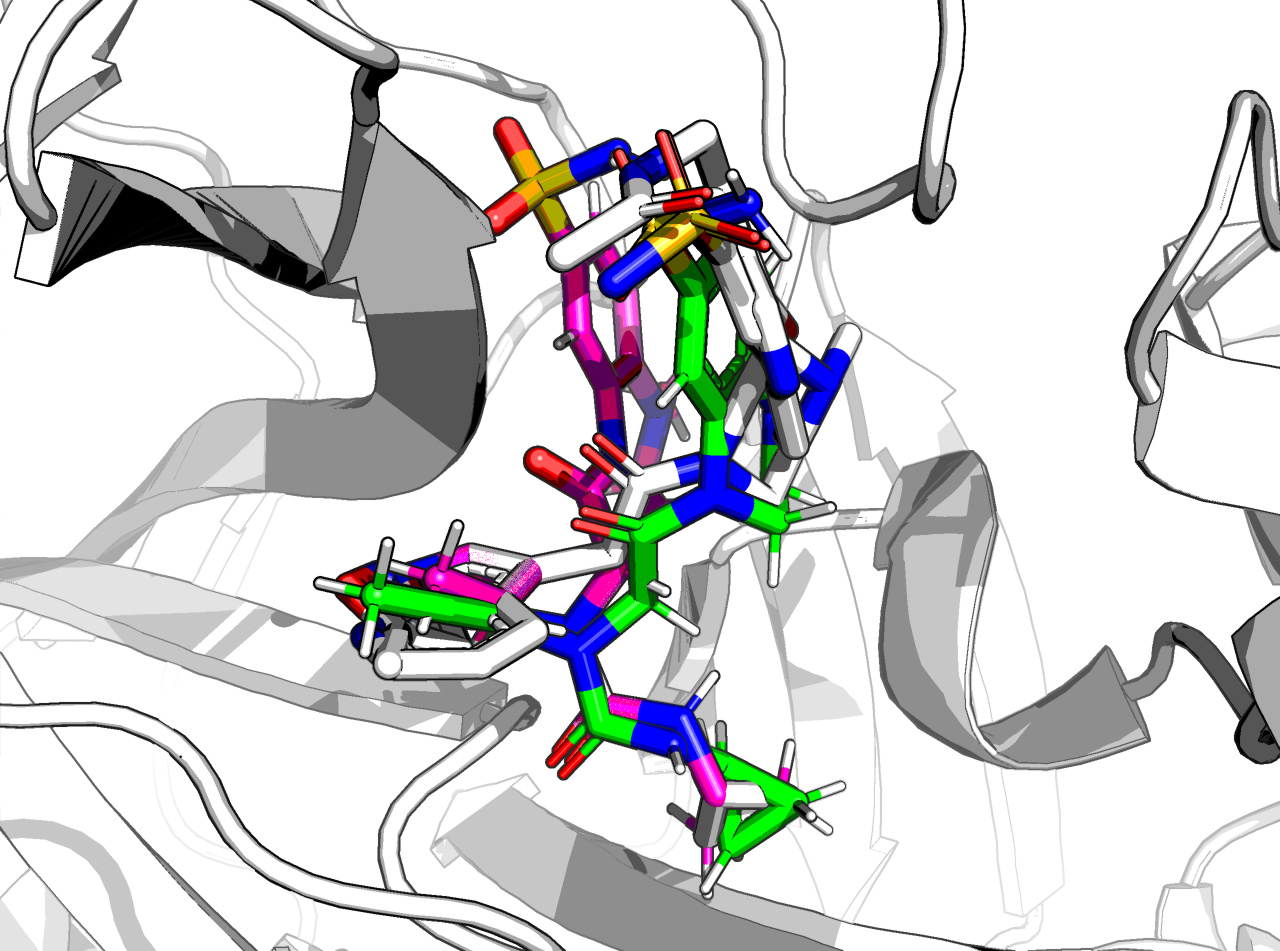
Even if an aromatic + non-aromatic ring is morally wrong (THC has one too hence its illegality) it minimises fine, with some strain on the N-methyl due to the positional constraint due to minimisation.
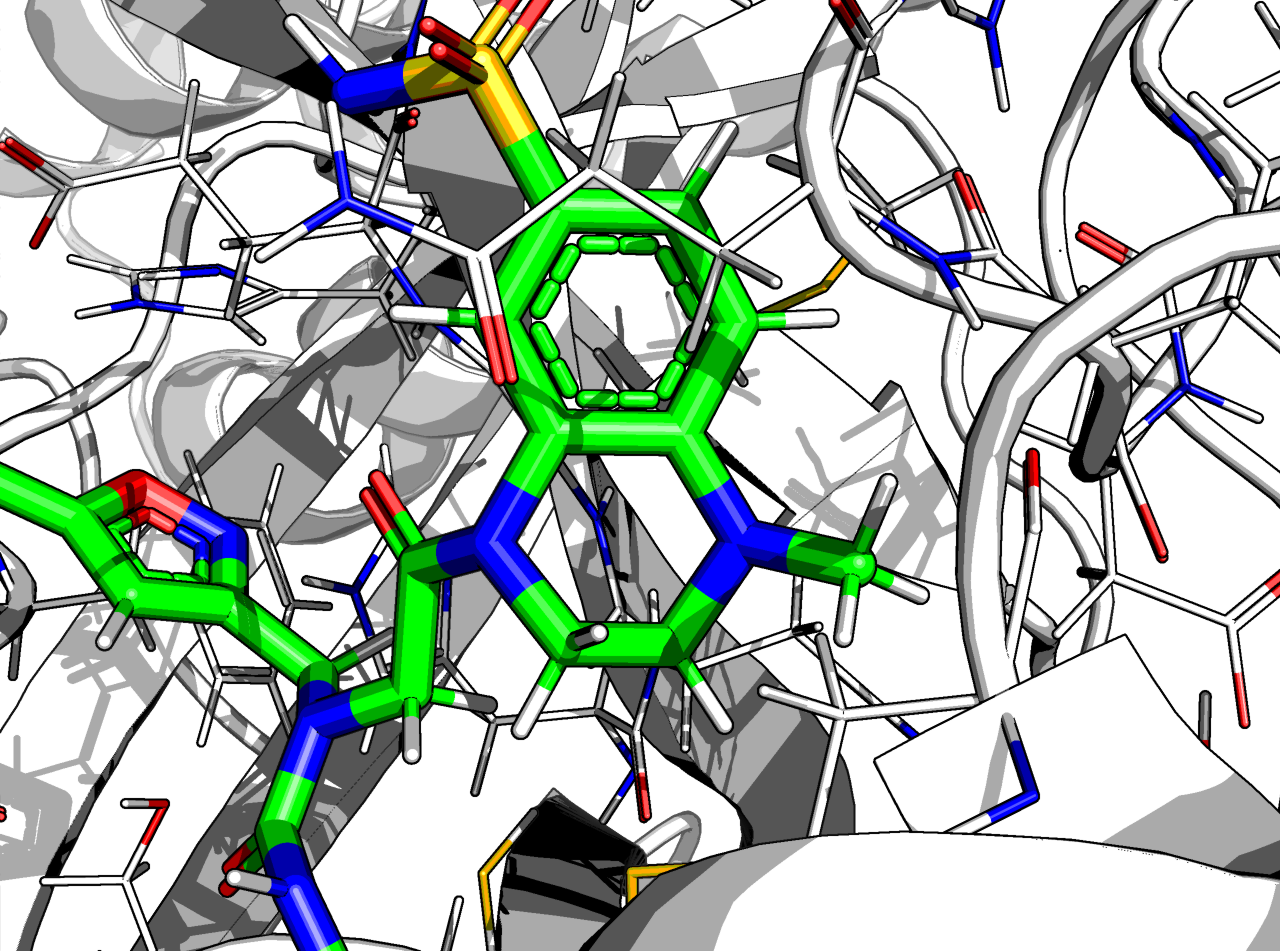
The combined RMSD is 0.58 Å, the ∆∆G_bind is -11 kcal/mol (Rosetta ref2015 FF) and has 32 heavy atoms mapped, so it’s a happy molecule.
- The individual RMSDs to the three hits are roughly equal.
- The solvatation term is very unhappy in the unbound state (for all three), but it is still not fully happy though, I believe because of the space in front of the ring —the S-delta of M49 is 5.9 Å away— but this is common to all three submissions.
I have no idea why the the cyclopropane rotates around.
FRA-DIA-3f9ae552-1x.holo_minimised.pdb (424.8 KB)
FRA-DIA-3f9ae552-2
This time too much is ambiguous and the mapping is subpar. So I did the following surgery.
from rdkit.Chem import AllChem
from rdkit import Chem
hit_codes = ['x0104','x0397','x0946','x1093']
hits = [get_mol(i) for i in hit_codes]
for h in hits:
print(h.GetProp('_Name'), Chem.MolToSmiles(h))
# x0104 CC(=O)NCCc1c[nH]c2ccc(F)cc12
# remove the amide sidechain.
hits[0] = AllChem.DeleteSubstructs(hits[0], Chem.MolFromSmiles('CCNC(=O)C'))
# x0397 Cc1cc(CN(C)C(=O)NC2CC2)no1
# is nice.
# x0946 NS(=O)(=O)c1ccc(Br)cc1
# remove the amide sidechain.
hits[2] = AllChem.DeleteSubstructs(hits[2], Chem.MolFromSmiles('c1ccc(Br)cc1'))
# aminosulfonate is 2.5 Å away from the tip of the indole. Fragmenstein in unmerge mode has a Cutoff is 3
# x1093 CN1CCN(C(=O)Cc2c[nH]c3ncccc23)CC1
# remove both rings
hits[3] = AllChem.DeleteSubstructs(hits[3], Chem.MolFromSmiles('c1c[nH]c2ncccc12'))
hits[3] = AllChem.DeleteSubstructs(hits[3], Chem.MolFromSmiles('N1CCNCC1'))
# keto is 2.9 Å away from the side of the indole. Under the cutoff by a nothing
for h in hits:
Chem.SanitizeMol(h)
Victor.fragmenstein_merging_mode = 'none'
# this method is just one that calls Victor, which is the pipeline, by adding Mpro specific details.
reanimate(name = 'FRA-DIA-3f9ae552-2x', hits = hits, smiles='Cc1cc(CN(CC(=O)c2cc(F)cc3c2N=C(S(N)(=O)=O)C3)C(=O)NC2CC2)no1')
The combined RMSD is 0.8 Å (vs 31 atoms), due to all hits really, and the ddG_bind is -13.5 kcal/mol.
FRA-DIA-3f9ae552-2x.holo_minimised.pdb (423.1 KB)
FRA-DIA-3f9ae552-3
Same as the above. But x0305 was an inspiration. So I added it (but it still drifts off).
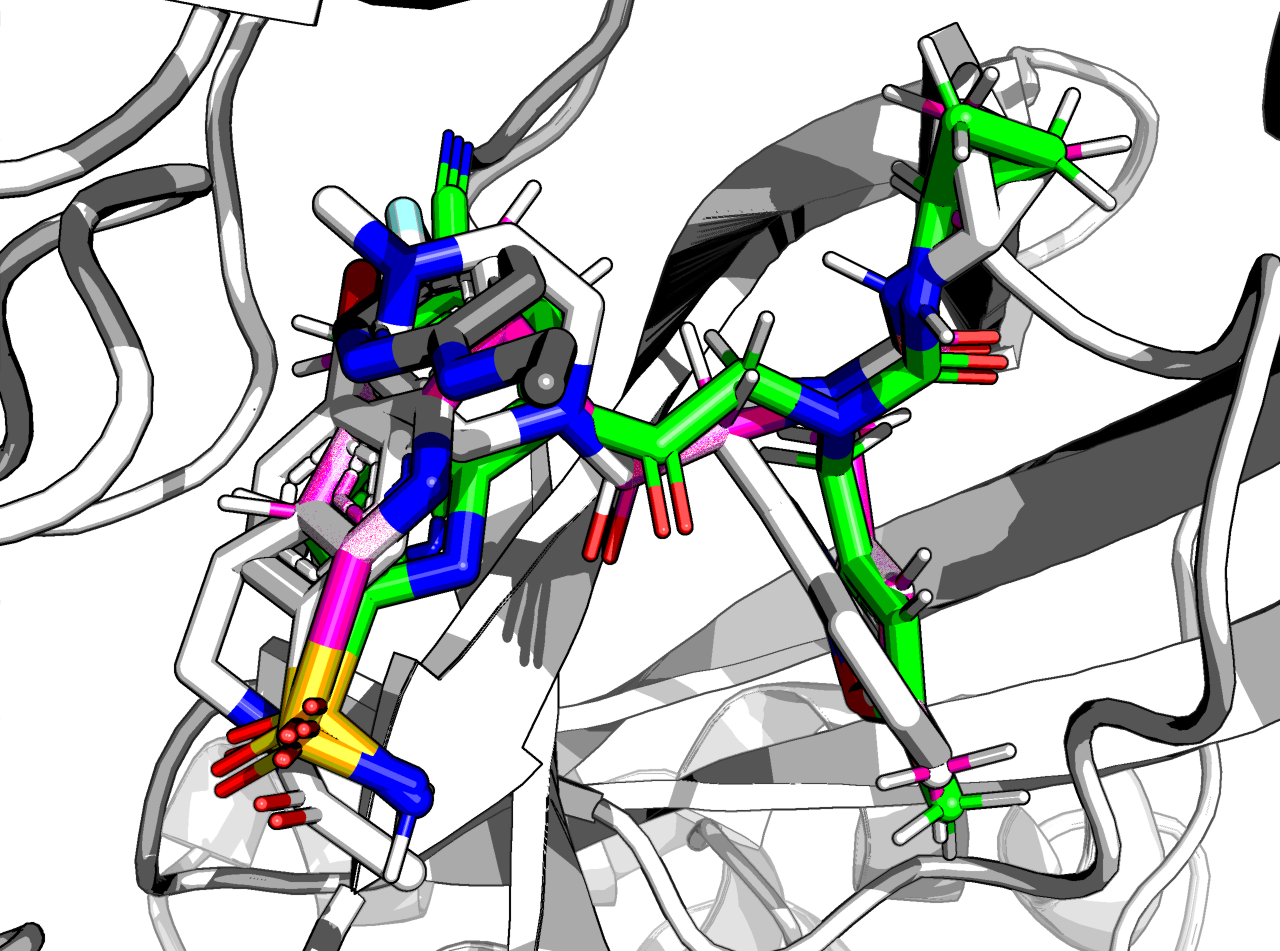
The nitrile is not quite where it should be, though:
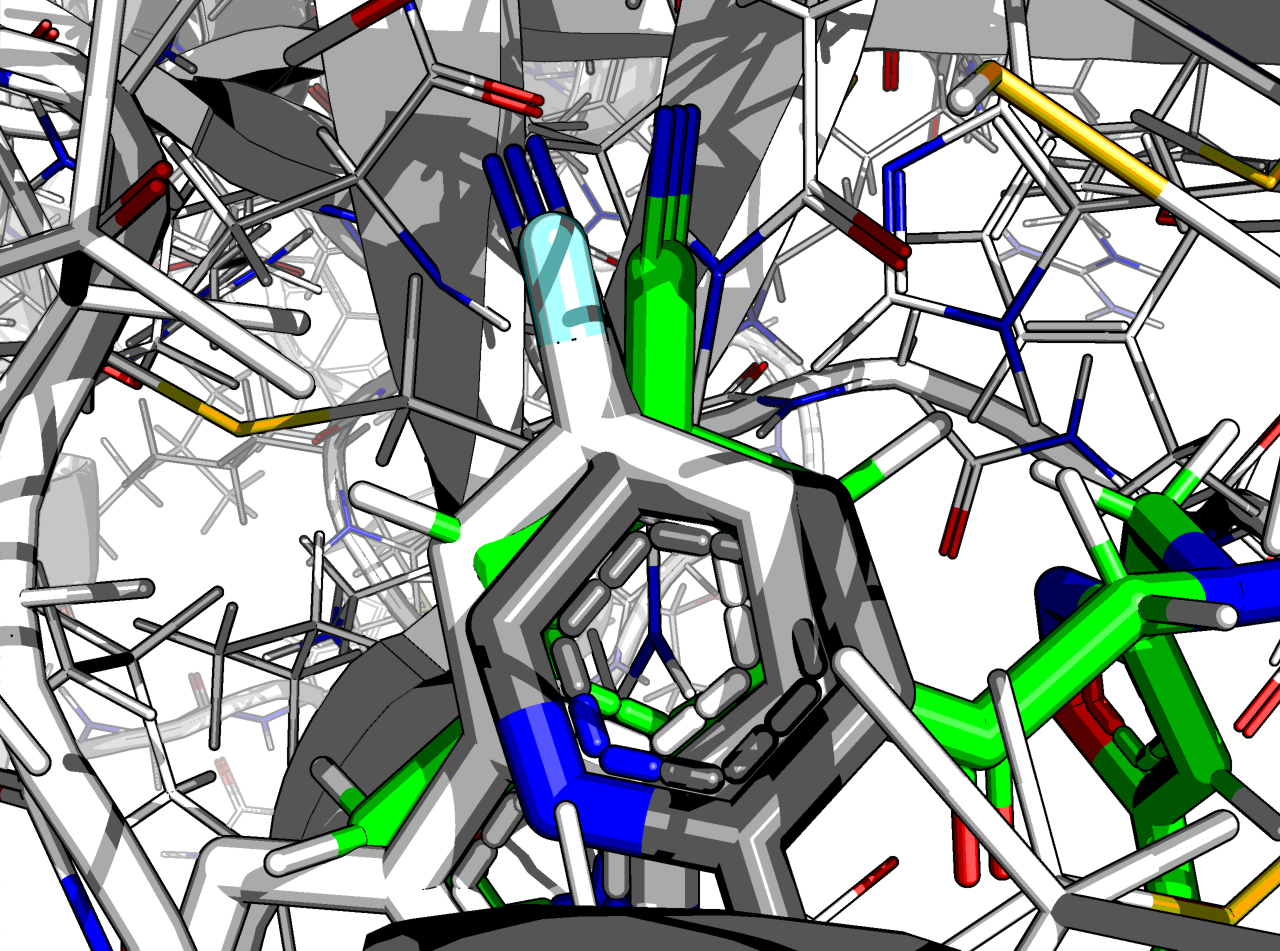
The RMSD is 0.8 Å and the ∆∆G_bind is -9.8 kcal/mol.
FRA-DIA-3f9ae552-3x.holo_minimised.pdb (423.7 KB)