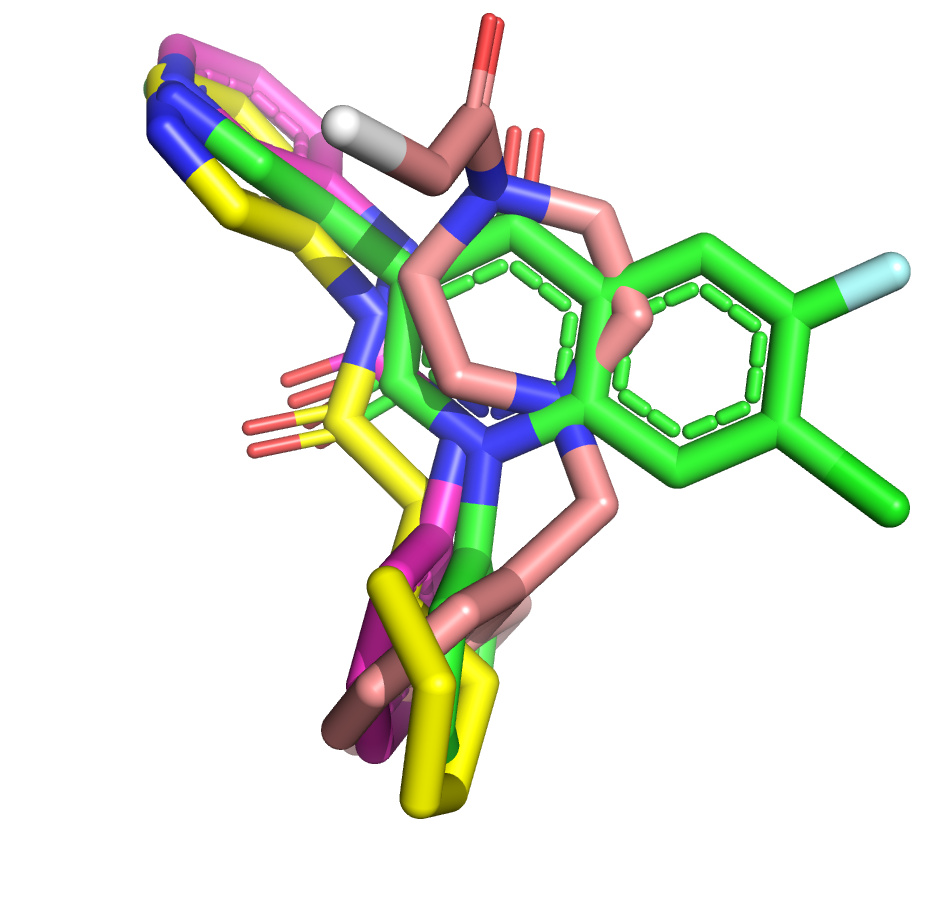
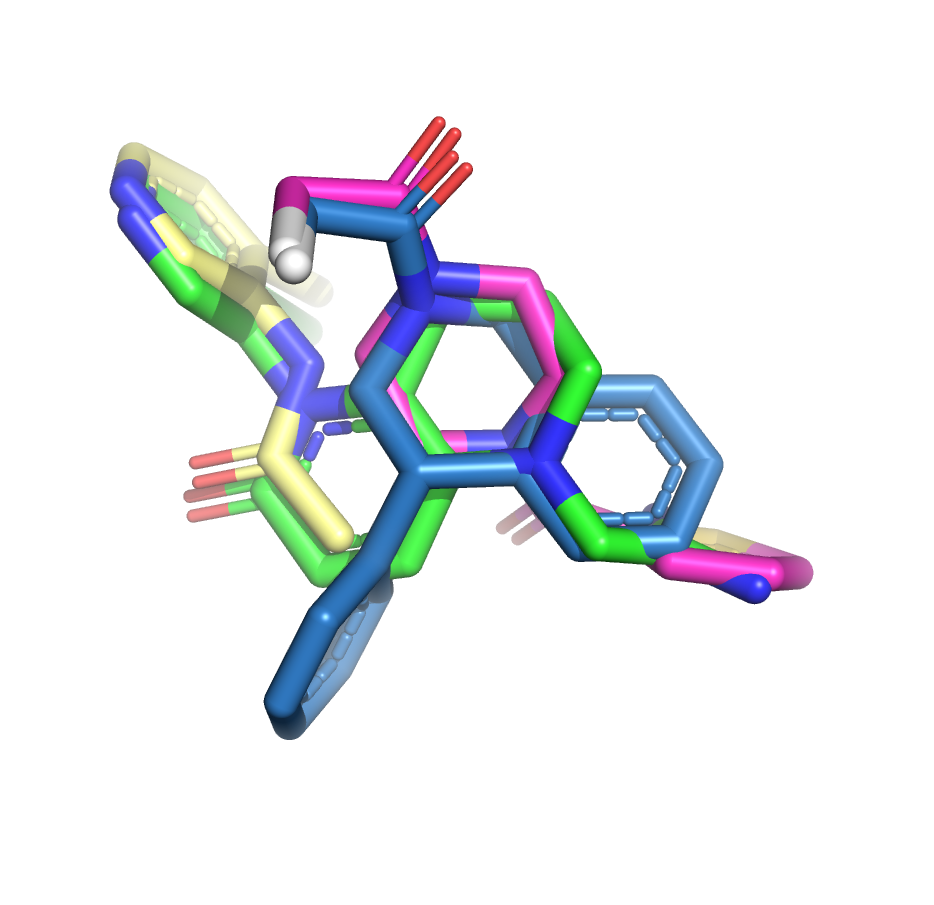
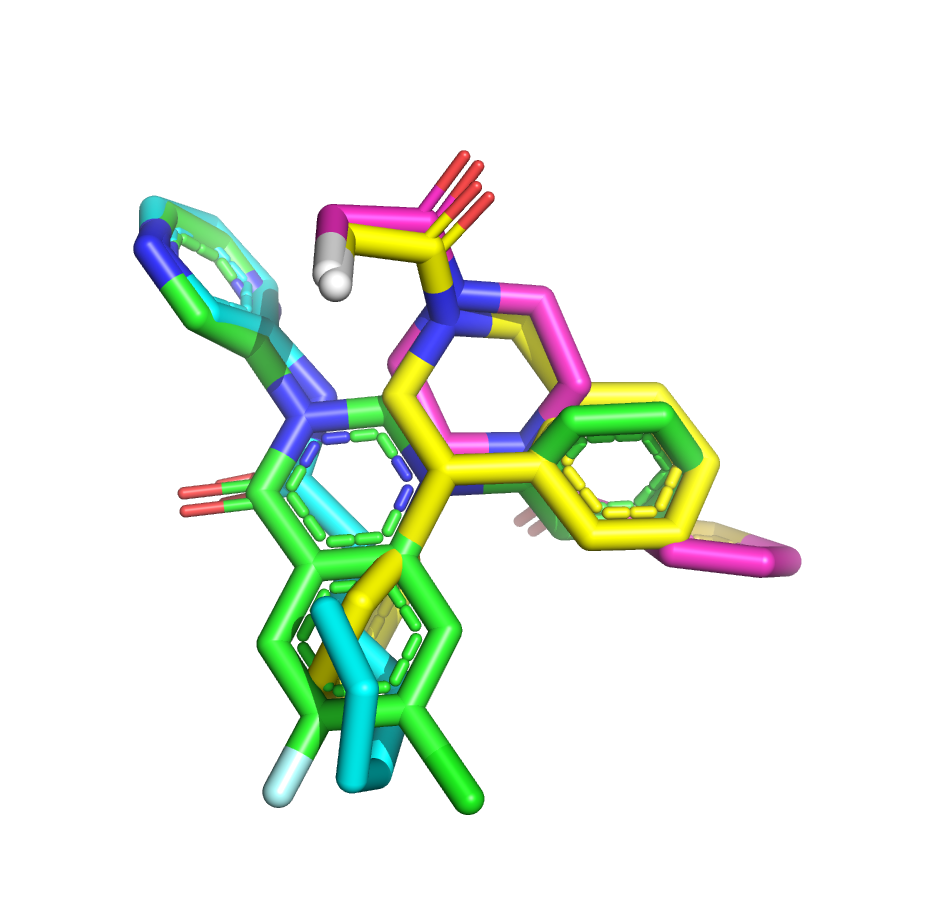
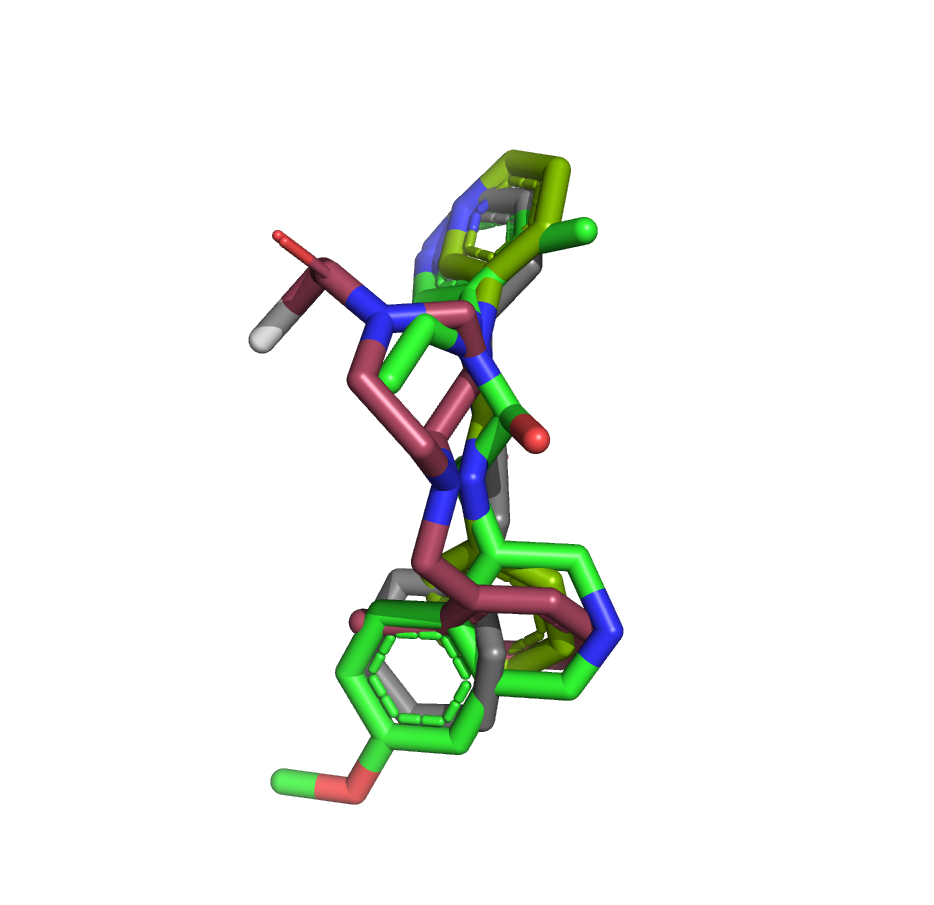
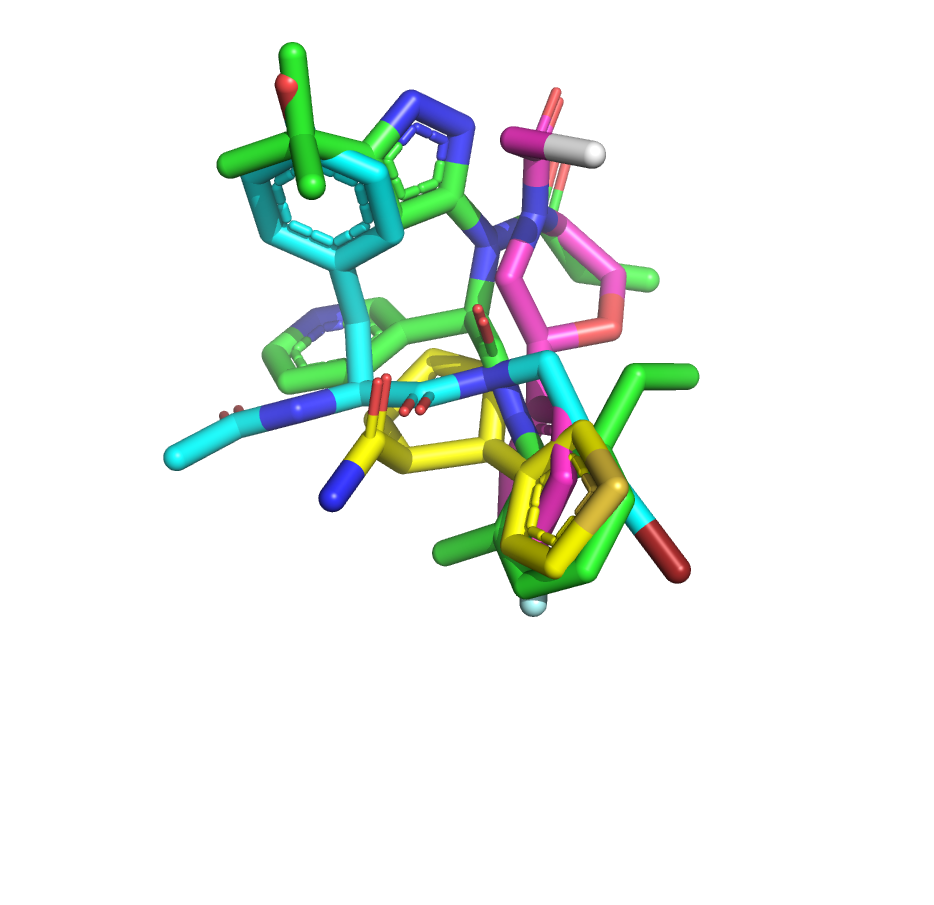
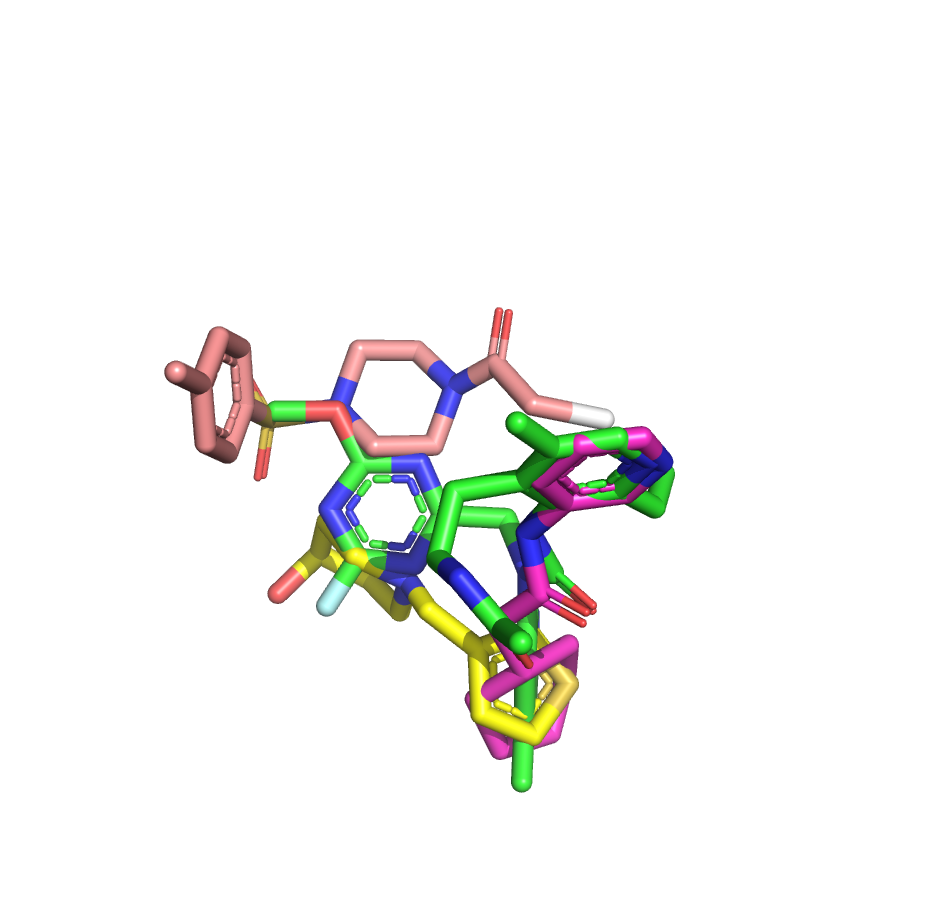
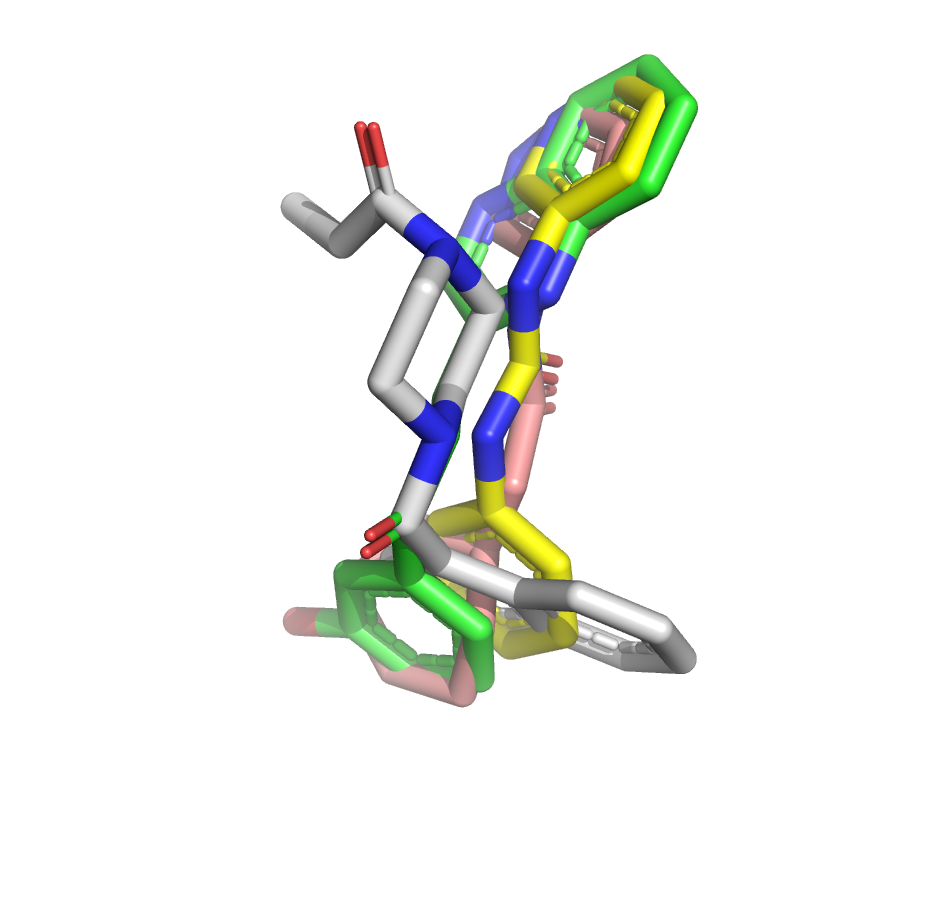I’ve taken another stab at the fragment merge scoring problem by implementing a simple script to identify the minimum fragment set that provides maximum coverage of docked molecule heavy atoms. The idea is that, for each hybrid docked molecule, we first identify the fragment with the largest number of heavy atoms within 0.6 A, then the next fragment with the largest number of heavy atoms within 0.6 A of any yet-unmatched atoms, and so on. We require at least three heavy atoms from each fragment to consider including it in the set of unique overlapping fragments.
The resulting list is sorted by the number of unique overlapping fragments, and produces some pleasing fragment merges from the moonshot compounds that float to the top (green is always the docked Moonshot compound, other colors are fragments):
The resulting SDF file—with listed overlapping_fragments—is here:
A corresponding CSV file is here:
These files also contain a score of the (fractional) number of heavy atoms that overlap with any fragment heavy atoms (within 0.6 A) and the total ligand volume, which may be useful as well.
I’ll see if these can be reformatted for Fragalysis upload, and will also investigate whether we can score very large synthetically accessible libraries by combining this with OE FastROCS searches.
Tagging @Waztom since @frankvondelft suggested he may be able to continue to refine this approach to prioritize designs that produce pleasing fragment merges. Also tagging @andrea who may have other good ideas about how to score designs for overlap with multiple fragments.
About the scoring, here are some thoughts. For Fragmenstein which also does a fragment merging based on position (probably very similarly*), the in-protein minimised followup molecule was scored simply with the un-aligned RMSD calculated from the concatenated set of contributing atom pairs between each hit and the followup (github.com/matteoferla/Fragmenstein#mrmsd), meaning that atoms from the followup that mapped to different hits were score more times. There was a discussing of using the RMSD of the merged scaffold only (without the novel bits in the followup) vs. the placed, but @frankvondelft rightfully pointed out that an atom whose position is dictated by multiple hits should be weighted more. There is probably a mathematically more elegant solution, which would be really cool.
Also probably worth mentioning is the dead-end avenue of B-factor weighting, I was interested in the B-factors of the hits, which would be nice to be used as non-linear inverse weights, but they are probably not consistent between structures and apparently the XChem pipeline does not always give them due to the manual steps. **
I did not use anything more sophisticated as I am not too knowledgeable on the topic and I was worried about behind the scenes alignment, eg for SuCOS, I have not tested whether a molecule translated by N Å scores differently than one that isn’t.
(* but with a cutoff of 2 Å, which as no atom can be present in two sets works just as well as .6 Å except for issues with 5-ring to 6-ring, which are fixed by the minimiser (Egor))
** EDIT: the logic behind the interest in the B-factors is that sometimes the hits stick out of the protein without any interactions and it would be nice to capture that. Here is an example (ignore the JS spiel it was for a feature suggestion that would not have worked).