One question of potential interest to COVID Moonshot designers is whether the potency benefits of isoquinoline at P1 relative to pyridine are maintained when the P1 heterocycle is linked by CH2 (as opposed to NH). In terms of potency, an isoquinoline at P1 needs to ‘pay its way’ since it’s bicyclic (naphthalene is less aromatic than benzene and therefore more reactive) as well as being bulkier and more lipophilic than pyridine. Here is some SAR analysis which suggests that isoquinoline at P1 may be less beneficial (relative to pyridine) when linked to carbonyl by CH2. Let me know if anything is not clear and/or if you spot any errors. This analysis has implications for lipophilicity management in what I’ll call the ‘benzotriazole series’ (isoquinoline has been substituted for benzotriazole in this series) and I’ll mention @mc-robinson @edgriffen @alphalee.
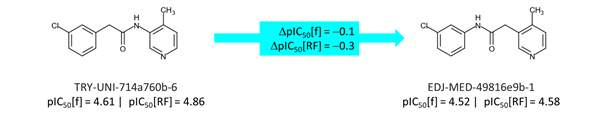
The starting point for the analysis is to note that ‘reversing’ the acetamide linker has a minimal effect on potency (f: fluorescence; RF: RapidFire) for methylpyridine at P1.
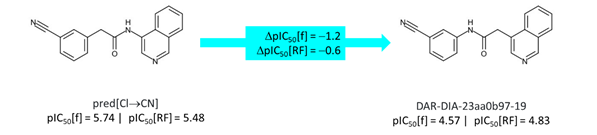
While ADA-UCB-6c2cb422-1 has been assayed, the other compound required for making the comparison for quinoline at P1 has not yet been synthesized although it was submitted as PET-UNK-8922bd3c-1 (and has been ordered). However, the ‘reversed acetamide’ DAR-DIA-23aa0b97-19 has been assayed and I’ll mention @Daren_Fearon who designed it. The isoquinolinyl amide with 3-cyanobenzyl at P2 has not been synthesized although its potency can be estimated as shown below.
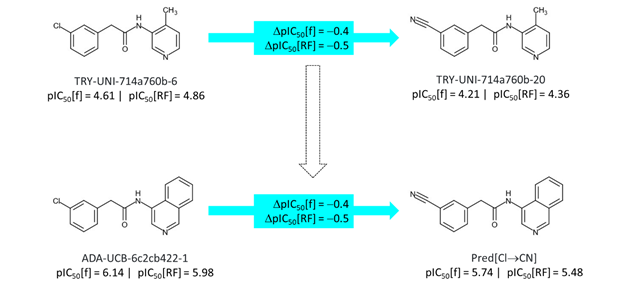
The estimate for the effect of ‘reversing’ the acetamide linker can be derived as shown below. While ‘reversing’ the acetamide linker of TRY-UNI-714a760b-6 results in a small decreases in potency, applying the same structural transformation to ADA-UCB-6c2cb422-1 is predicted to result in a 1.2 log unit decrease in potency in the the fluorescence assay. This suggests that a significant proportion of the potency benefit (relative to pyridine) of the P1-isoquinoline will be lost when the acetamide linker is ‘reversed’.