In recent decades the pharmaceutical industry has not favoured covalent ligands. Perhaps this is why most virtual screening studies have ignored that possibility.
I have demonstrated to my satisfaction that it is possible to dock a variety of covalent molecules with standard warheads as well as a few novel ones. This includes molecules which bridge the CYS(145) HIS(41) gap etc as well as interact elsewhere in the binding site. The scoring functions should be better than “selection by eye” but do not attempt to address the reversibility of the binding nor do I have any reasons to beleive they will perform better than non-covalent scoring functions.
Some of the molecules I’ve docked are similar to previous submissions but many are either from commercially available screening libraries, virtual libraries or dream libraries (where the chemistry required eg for heterocyclic ring closure is in principle possible but not trivial).
I’m not sure what selection process would be best. Partition into subsets based on other residue interactions or by electron withdrawing/donating properties of groups connected to the warhead (which have the potential to impact the kinetics). An alternative approach would be molecular properties …
Thoughts and suggestions are welcome!
1 Like
This is why the focus was dropped from my limited understanding. But it was addressed.
In March-April I docked in Rosetta the covalent submissions and @garrett has since done the same in Autodock 4. And indeed, all combinations of the five main warheads discussed were tried. However, the problem was not the covalent docking or modelling of the reaction steps to calculate the energy barrier as discussed with @dmoustakas —although admittedly I started and didn’t bother analysing it as it was not of interest anymore.
From what I can tell the reason why covalent submissions have been deprioritised is one the med-chemists have discussed at length, namely the strong concerns of toxicity of the warheads based on a list of previous leads making it to market. Even if I am a computational biochemist and not a chemist, so not at all qualified to comment on this, given that this project has a real time factor (Covid), avoiding to try less proven classes of molecules makes perfect sense to me.
Of course, there are many really good covalent drugs and https://www.news-medical.net/news/20200416/Gold-containing-drug-auranofin-kills-9525-of-SARS-CoV-2-virus-in-48-hours-in-lab-conditions.aspx (which is old news) would suggest that there is good potential such drugs in the treatment of Covid-19.
Toxicity (and less serious side-effects) are a real problem in pharmaceutical research but that isnt really a surprise since so little effort is put into avoiding it in the lead generation, lead selection and lead optimisation stages.
I agree that it sounds like a self-fulfilling prophecy.
There is an ongoing discussion here Data Release and Updates: 2020-06-12 about the panels to test off-target binding and direction.
Reversibility is an issue that needs to be considered for covalent inhibitors of cysteine proteases. These notes may be of interest:
I remain intrinsically unhappy making subjective selections of a handful of molecules from thousands or tens of thousands of molecules and today’s scoring functions are no better than those of 20-30 years ago - roughly as good as visualisation of the hits! I had some success a few years ago using a panel of P450 structures to filter out molecules and leave better drug candidates. It’s going to take some time (well at least another week or two) to configure and test a panel of human cysteine proteases for virtual counter screening for covalent and non-covalent ligands. I’ve no doubt that this would eliminate some viable molecules (due to false positives on the counter screen) but I would expect the numbers of molecules left to continue to be much larger than the synthesis capabilities.
Some people have argued that when purchasing commercially available molecules it’s best to test as many as possible for activity and then eliminate those that show activity against a counter screen of other assays. However, purchasing and testing costs are much lower than synthesis costs.
It has been suggested by L Cianni et al in J Med Chem 2019 62 (23), 10497-10525 that selectivity for non-covalently bound inhibitors for Cysteine Proteases is generally unachieveable and this is reflected the lack of clinical success for such compounds.
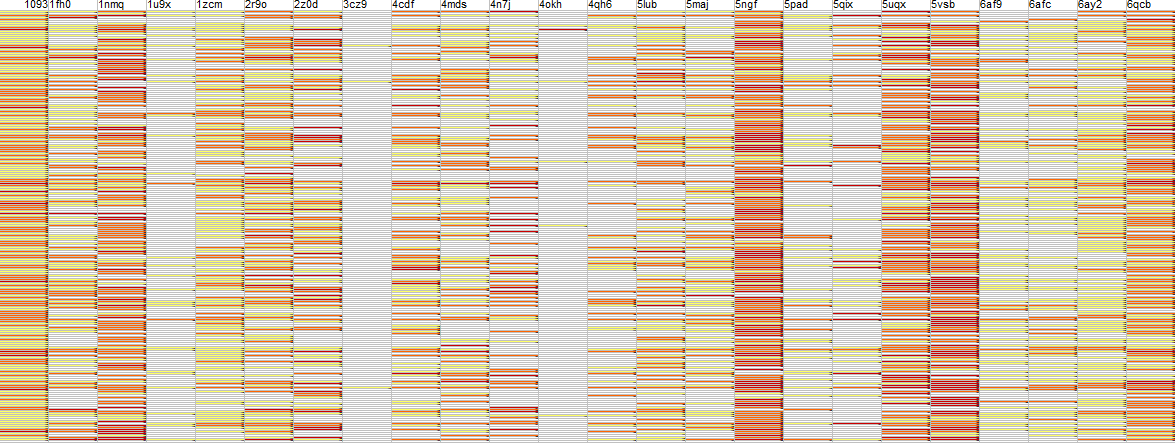
I took a representative 1000 molecules which are commercially available spanning a range of properties including flexibility and docked them into 5RF7 using our THINK software giving approximately 200 hits (these have not been submitted as inhibitor designs). These were then counter-screened against an arbitrary panel of over 20 human proteases. The summary provisional results are shown in the image.
The colour coding is: white not a hit; yellow weak score; amber moderate score and red very good score. Each row is a different molecules and each column is a different protein with 5rf7 the first column and a horizontal white line would indicate complete selectivity for that hit. It is immediately apparent that complete selectivity is problematic especially against some human proteases (Caspase 1nmq; Ubiquitin 5ngf, 5uqx & 5vsb; Cathepsin D
6qcb).
I have failed to find a rationale for the choice of SARS-Cov-2 Mpro as a target in this project and I was wondering how it compares to alternatives. These authors (and others) advocate that only covalently bound ligands have any reasonable chance of clinical success. Should we therefore eliminate all non-covalent ligand from purchase, synthesis and testing programmes?
I think the main difficulty with non-covalently-bound inhibitors of cysteine proteases is achieving adequate affinity with acceptable physicochemical properties. I think this the primary reason that industry has tended to favor peptidomimetics with ‘reversible’ warheads in cathepsin programs. Generally one would not expect to get selectivity from the covalent bond because the warhead binding modes tend to be very similar for different cysteine proteases. My working assumption is that selectivity for reversibly-bound covalent inhibitors of cysteine proteases comes primarily from the non-covalent interactions. This blog post may be of interest
I agree that selectivity for both covalent and non-covalent inhibitors has to rely on differences in the binding sites such as exploting non-covalent interactions. I would argue that a potential issue with non-reversible inhibitors is that they may never reach their intended target (if the inhibit every cysteine-protease they encounter).
20+ years ago, pharma research focused on potency first and then optimised a series for acceptable physiochemical properties. Post Lipinski, it’s been tempting to filter out too aggressively on properties and functional groups with consequential delays finding sufficient potency for Mediciinal Chemistry to start.
IMHO there’s not enough data released on why clinical trials fail. Looking at the human cysteine protease crystal structures the extent of conformational change is a potential concern as this might undermine any rational approach to selectivity. Obviously, without selectivity it is doubtful whether a covalent or non-covalent Mpro inhibitor would be a viable clinical trial candidate.