This post contains results of a docking model that was optimized using both the crystallographic fragment screening data, as well as the biochemical screening data published (5/7/2020) for ~120 compounds. This structure of SARS-CoV-2 Mpro used in this model was selected to optimize early recovery of known actives. Summary of the validation data is shown at the end of the post.
Overview of selected hits
2020-05-15_ranked_submissions_model_02084_FORMATTED.sdf (802.3 KB)
submission_02084.zip (158.5 KB)
This file contains a ranked list of 200 compounds, sorted by docking score (lower is better), and formatted in Fragalysis v1.2 format. The file submission_02084.zip contains the docking receptor in Fragalysis format. The SD file contains 3D coordinates, posed into the the following docking receptor, uploaded in the following links in both PDB format and in OEDocking receptor grid format:
DOCKING_MODEL_02084_COMPLEX.pdb (730.0 KB)
DOCKING_MODEL_02084_RECEPTOR.pdb (744.5 KB)
DOCKING_MODEL_02084_OEDOCKING.oeb.gz.zip (850.7 KB)
The complete set of docked poses (3649 molecules), containing 3D docked coordinates and a docking score (“Fred Chemgauss4 score”) is contained in the following file:
ALL_DOCKED_POSES_MODEL_02084.sdf.zip (2.9 MB)
@JohnChodera
All poses included here for the benefit of retrospective analysis and comparison of performance across multiple methods. It’s worth noting that this total set of all posed compounds contains both non-covalent and covalent designs.
Receptor Preparation
An X-ray crystal structure of SARS-CoV-2 Mpro complexed with a covalent inhibitor (PDB 6LU7) was used as a starting point for docking model development. The N3 ligand occupies a large volume of the active site, in the hopes of yielding a docking model with less bias for any particular pocket in the active site, since the goal of many of the submitted designs was to link/merge fragments binding to different active site pockets. The 6LU7 PDB model contains the covalent adduct of the N3 inhibitor to the catalytic Cys145 residue, so the non-covalent complex was modeled using the MOE software to break the ligand-Cys bond, generate the pre-reaction ligand species, assign protonation states and partial charges, and minimize any clashes. This structure contains the deprotonated Cys145 thiolate species.
A molecular dynamics simulation of the non-covalent complex model was performed using the Desmond software, using the OPLS3 force field and an SPC water box. A 1 us trajectory was used to generate an ensemble of 5000 receptor conformations, from which the final docking model was selected using an exhaustive docking method. OEDocking/FRED receptor grids were prepared for each of the 5000 structures using the Spruce toolkit with default values, and a training set of ligands were docked into each structure. The structures that yielded the highest early enrichment factor were selected for docking. Details of the optimization process are described in the Retrospective Analysis section.
Ligand Preparation & Docking
Structures of all submitted proposed designs were downloaded, and were filtered to remove any structures containing greater than 7 rotatable bonds or MW > 600, leaving ~3700 compounds. The OpenEye toolkits were used to assign the single most likely protonation state at neutral pH for all of the compounds. The Omega2 program, running with the following parameters:
-ewindow 10.0
-maxconfs 50000
-strictstereo false
-flipper true
was used to enumerate unspecified stereocenters, as well as conformations, yielding a total of ~8.6K unique species and ~4.3M conformers.
The Fred program (OEDocking) was used to dock the multiconformer library against the receptor grid, using the following settings:
-dock_resolution standard
-hitlist_size 0
Docked poses were written out in SD format, containing a field with the Chemgauss4 score value for each pose. For each compound, the top-scoring of the enumerated stereoisomers was retained, yielding a reasonably normal distribution of docking scores:
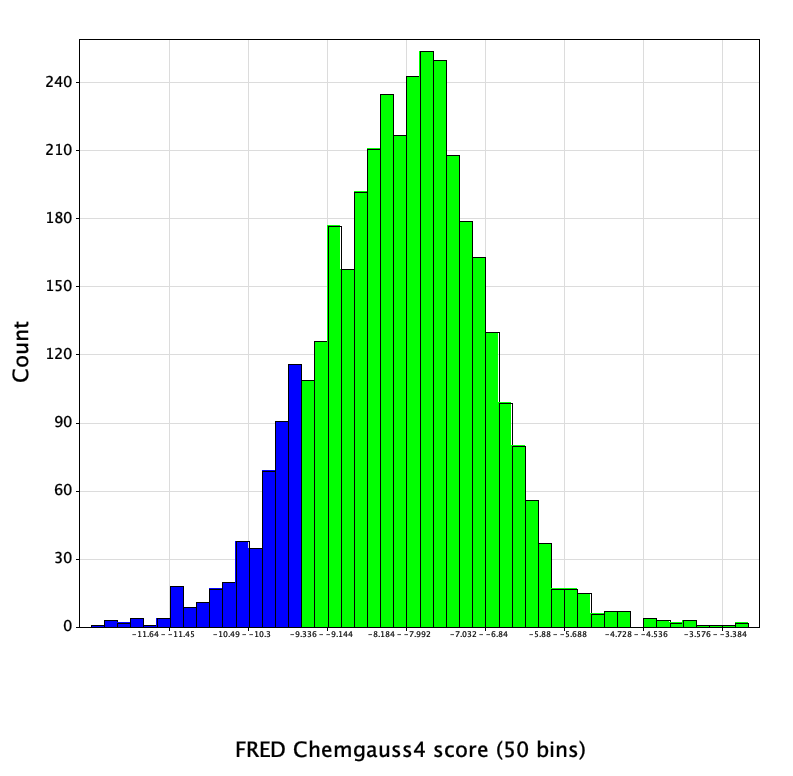
Since biochemical Mpro inhibition data had been published on 5/7/2020 for a number of non-covalent compounds contained in the overall set, I extracted the 12 actives from that preliminary data set and identified their docking scores in the following table:

Reassuringly, 6 of the 12 actives, representing 3 distinct chemical series, were found in the top-scoring tail of scores < -9.5 (shown in blue in the histogram above). Further increasing confidence in this model is the presence of a number of compounds scoring significantly better than the known references, which hopefully should increase the likelihood of identifying more hits of equal or greater binding affinity.
Hitlist Selection
The score distribution for the entire library had a mean value of -8.1, with a standard deviation of 1.2. 450 compounds with scores < -9.5 (greater than 1 SD below the mean) were selected (shown in blue in the histogram). Since the entire set of submitted compound designs contained both non-covalent compounds and those with covalent warheads, the compounds with warheads were removed, leaving 420 compounds in the set.
Visual analysis of this set revealed a large number of highly similar compound clusters, so a greedy clustering algorithm was used to filter out lower ranked near neighbors of higher ranked hits. The OEGraphSim toolkit was used to compute the tanimoto similarity (based on path fingerprints) for the first compound in the list to all lower-ranked compounds, filtering out all compounds with similarity greater than 0.7. The algorithm iterated in a greedy fashion through the ranked hitlist until all near neighbors were removed, leaving ~220 compounds, from which the top-ranked 200 compounds were saved and formatted into the Fragalysis V1.2 format.
Retrospective Analysis of Biochemical Data Set
The training set of reference ligands used for this model optimization were the non-covalent ligands assayed by Haim Barr at the Weizmann institute and published recently:
https://covid.postera.ai/covid/activity_data
Non-covalent ligands with < 8 rotatable bonds and < 600 MW were included in the docking (103 compounds):
2020-05-07_tested_ligands.sdf (232.4 KB)
2020-05-07_tested_ligands_CONFORMERS.oeb.gz.zip (1.1 MB)
The performance of each of the 5000 receptor conformation dockings was evaluated, using early enrichment as a metric to identify conformations that could be promising as docking models.
The best of these models yields an ROC curve shown below:
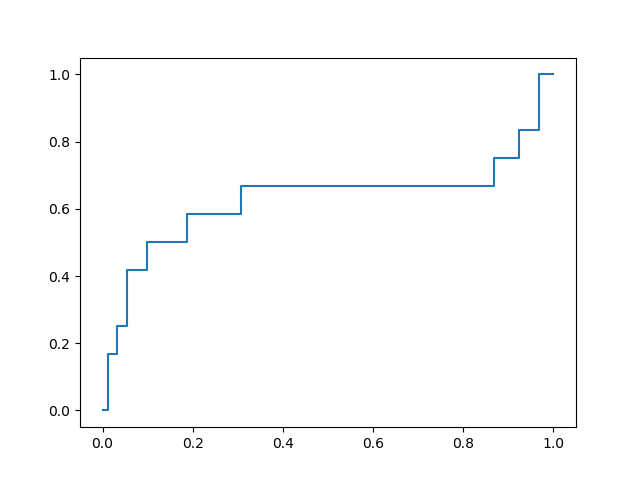
with a favorable early enrichment profile. By comparison, docking into the 6LU7 X-ray crystal structure yields the following ROC curve:
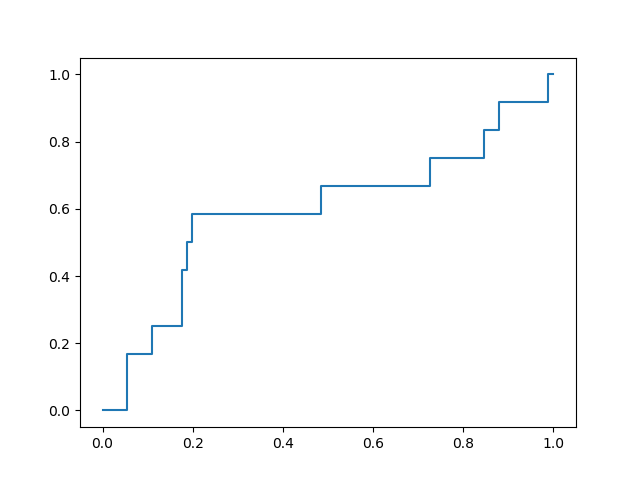
While not dramatically different, comparison of these two plots shows that the optimization yielded a docking model with double the rate of early enrichment than the X-ray structure provides.
This procedure yielded a receptor conformation with significantly improved early enrichment for this data set. Due to the similarity between this first data set and the remaining submitted designs, this receptor model was considered to be ideal for virtual screening in this context. Since significant early enrichment was found in screening the top 10% of the retrospective data set, I’m hopeful that the prospective docking selections, drawn from the top 10% of the submitted compounds (~400 of ~4000) should yield a favorable hit profile.
It’s worth noting here that the actives recovered in the docking from the first data set dock with poses that appear to be consistent with the expected binding modes based on the fragment crystal structures. For example, compound TRY-UNI-714a760b-22 binds with the following predicted mode:
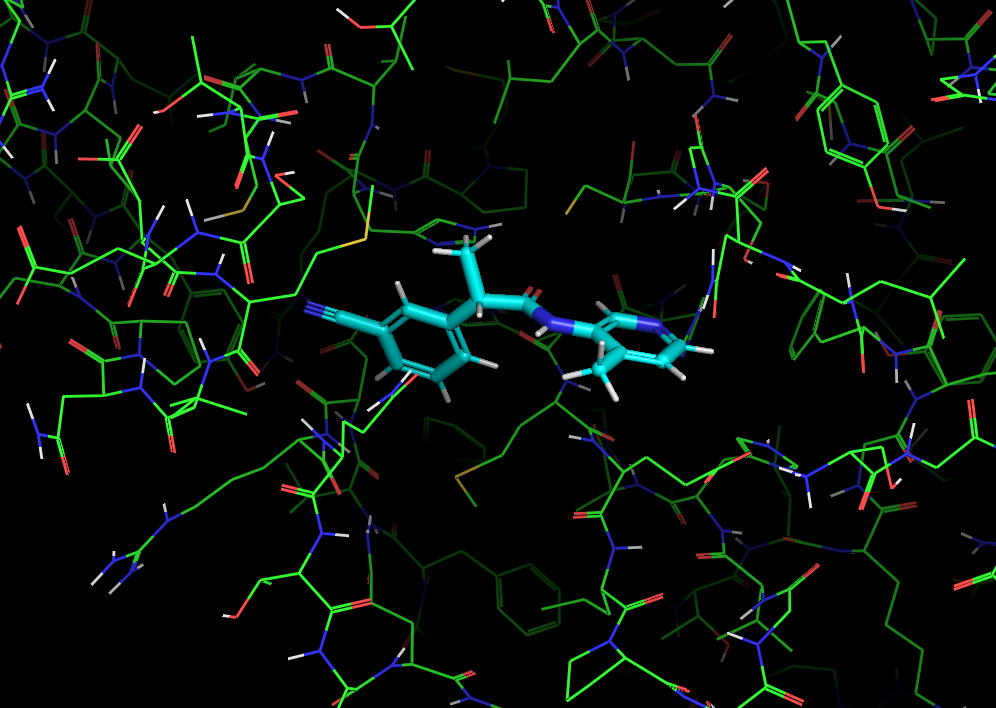
Which is similar to the binding of the nitrile-containing compounds x0305 and x1249, which also position the nitrile in the hydrophobic P2 pocket.
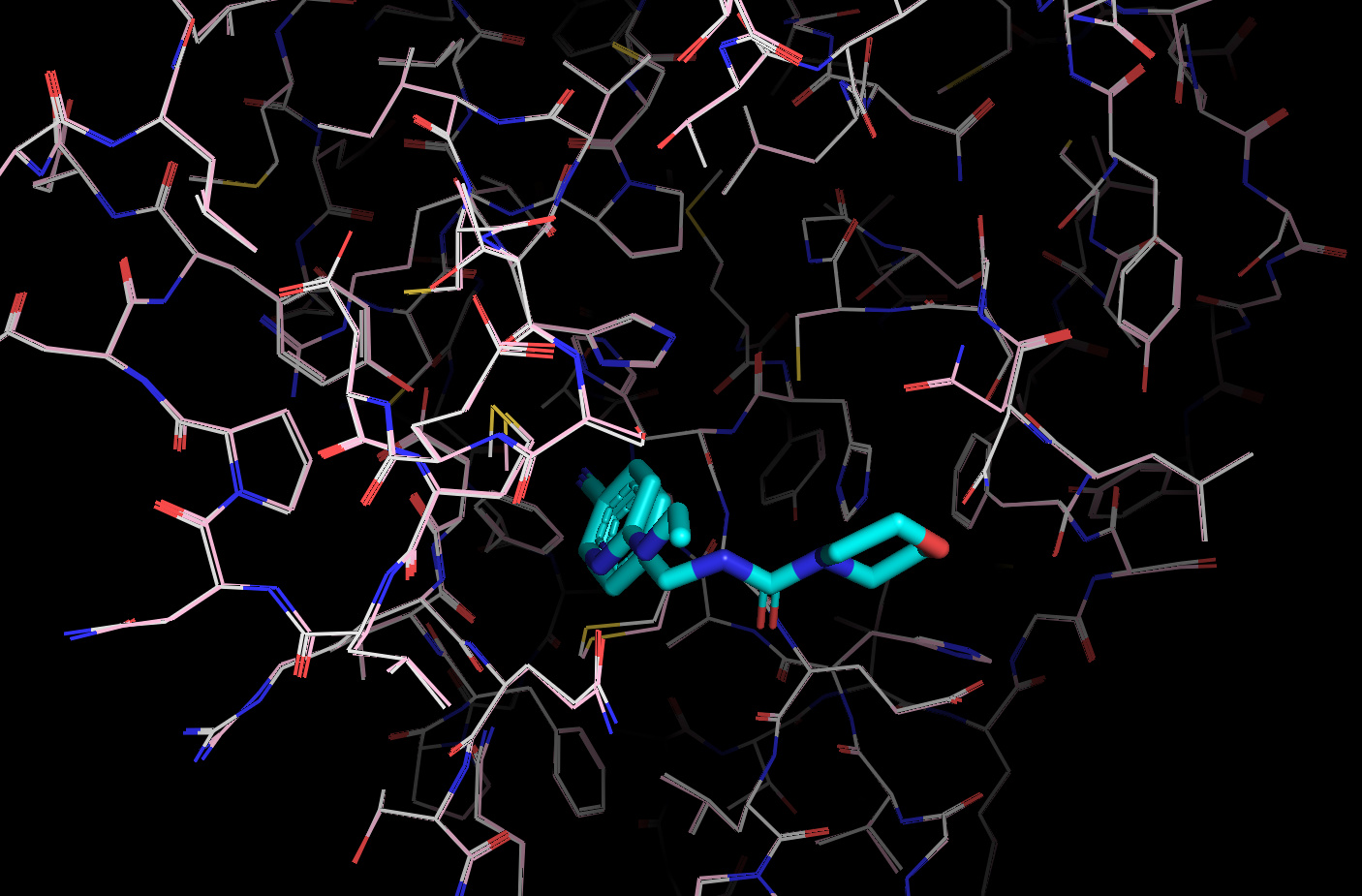
In general, this docking model/receptor conformation appears to favor compounds spanning the P2 -> P1 pockets, as exemplified by the Diamond structures x0434, x0678, x0967, x1093, x1249. This appears to be driven by the conformations of a number of key residues, including Asn142, Gln189, Met49, Cys44.
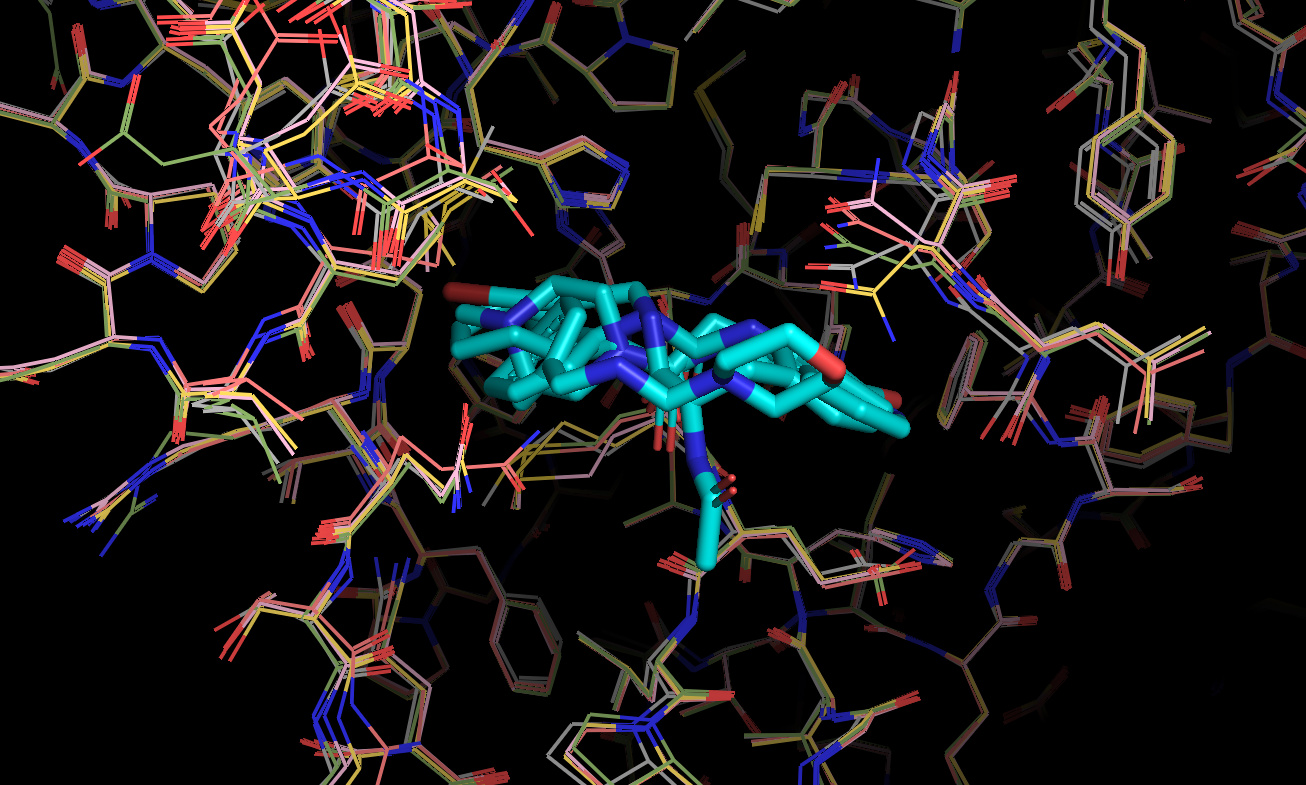
Therefore, this docking model/receptor structure may be ideal for designing/expanding on the these sorts of fragment structures.
