Hi all,
We’ve been busy collecting data! I’ll try to give an overview on recent progress.
In case you are wondering, I was just looking at the graph below, and we just yesterday hit the mark of over 500 compounds assayed. We also now have over 100 structures from Diamond UK of ligands bound to the protein.
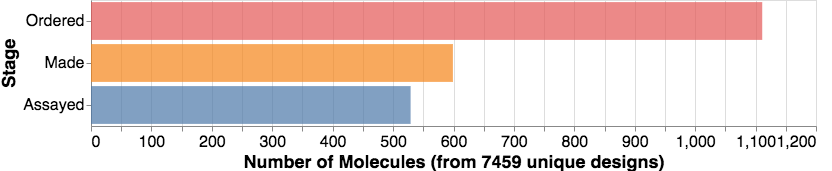
And as the graph shows, many compounds to come! Compounds are mainly being made at Sai Life Sciences (India), Enamine (Ukraine), and WuXi (China), then shipped for testing to Oxford, UK (Diamond UK + Oxford U.) and Israel (Weizmann Institute). This all shows the incredible international nature of this community, which also involves many of you submitting ideas (now over 7000 designs) from other diverse parts of the world.
Currently Promising Compounds
The compounds
@edgriffen created a very nice slide of 5 potential starting points so far (Tour of Learning from Docking, modelling and crystal structures), which I will shamelessly copy below. Note that the slide is slightly outdated. For example, we have since realized that the sulfone-piperazines with a chloroacetamide warhead are much more reactive than other piperazine-chloracetamides (I’ll explain more below).
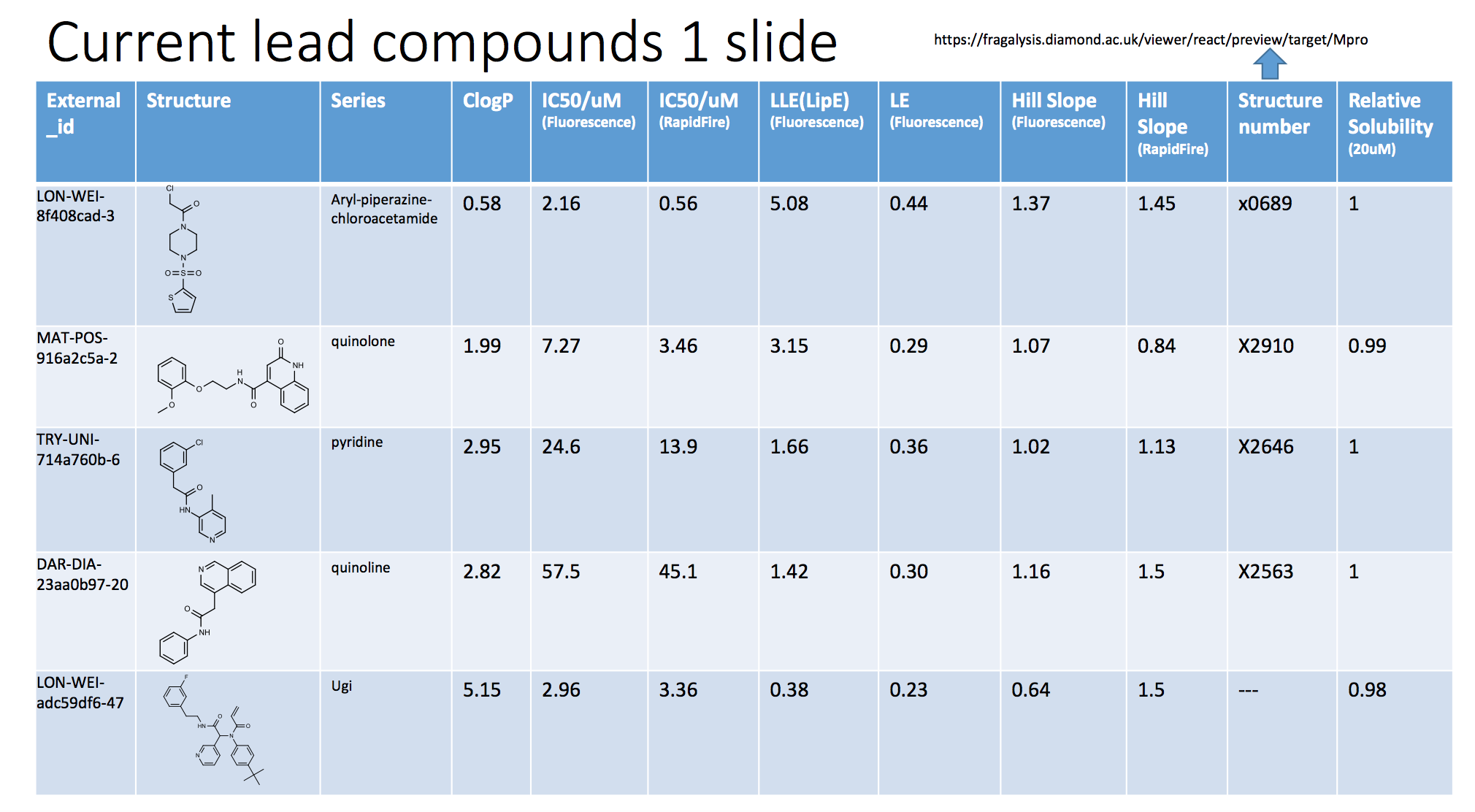
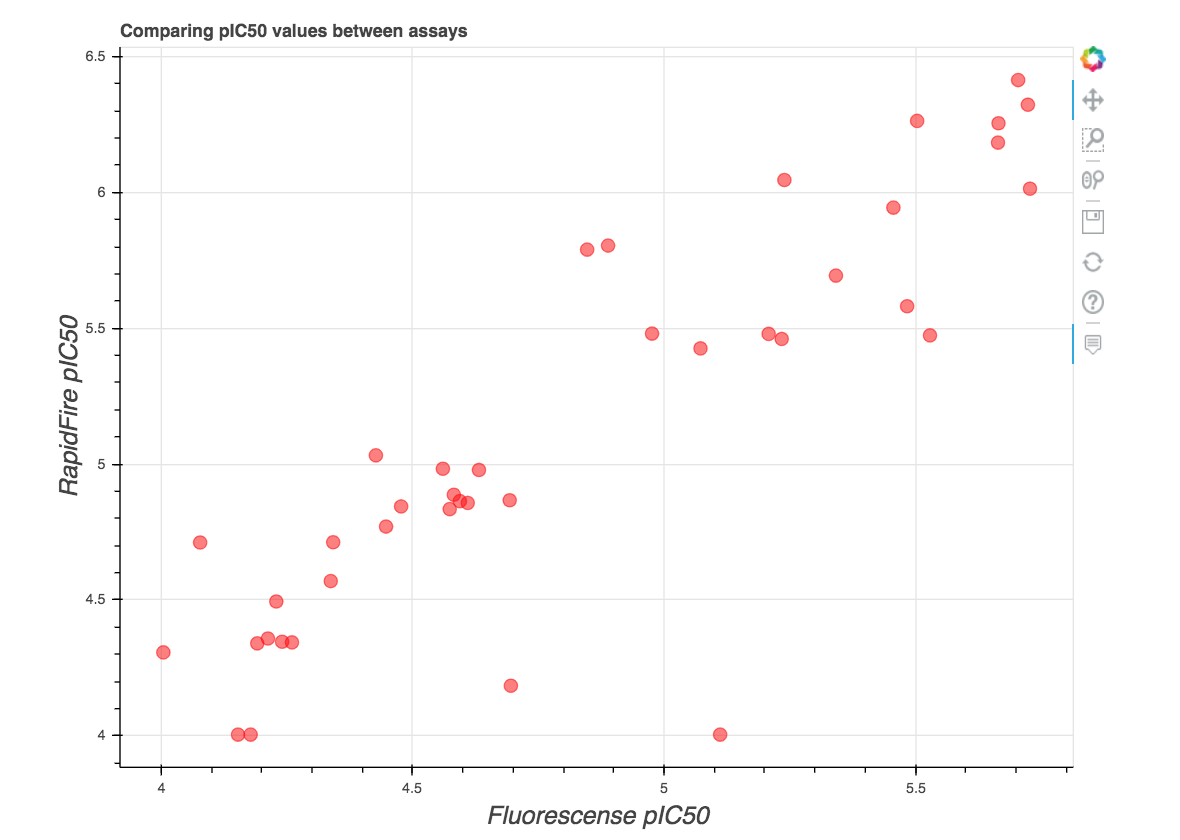
As you may notice, there are two IC50 values listed in the slide. This is because we now have assays up and running at both Weizmann (Fluorescense) and Oxford (RapidFire MS)! Even better, they generally tend to agree, as shown in this plot at https://covid.postera.ai/covid/activity_data:
Structures of these Compounds
As the slide also shows, we have structures on four of these compounds, and we do have an Ugi structure, just not the compound shown. @markc has posted an excellent overview of how these ligands fit in the binding pocket here Tour of Learning from Docking, modelling and crystal structures. I will again shamelessly copy some of the slides below.
First, Mark helpfully orients us in the binding pocket by using the structure of the N3 ligand (PDB: 6LU7) from https://www.nature.com/articles/s41586-020-2223-y_reference.pdf

an Ugi structure
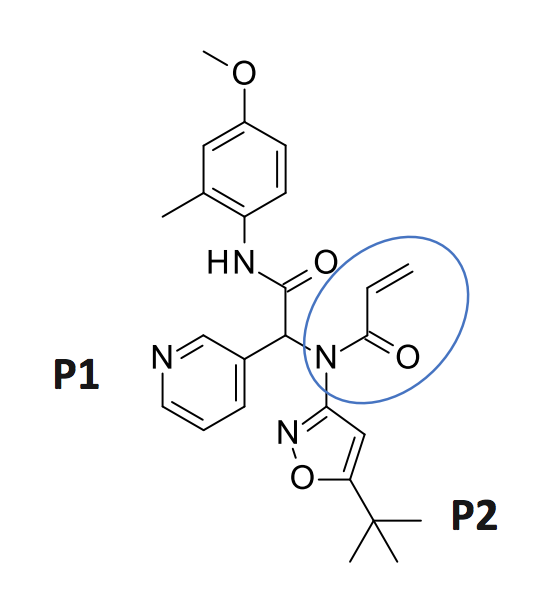
The first structure I’ll look at is an Ugi compound with an acrylamide warhead
LON-WEI-b8d98729-43, which is actually a pretty weak binder (23% inhibition at 50 uM). @MarkC provides a nice overlay of the structure (x2694 on Fragalysis) with the N3 ligand:
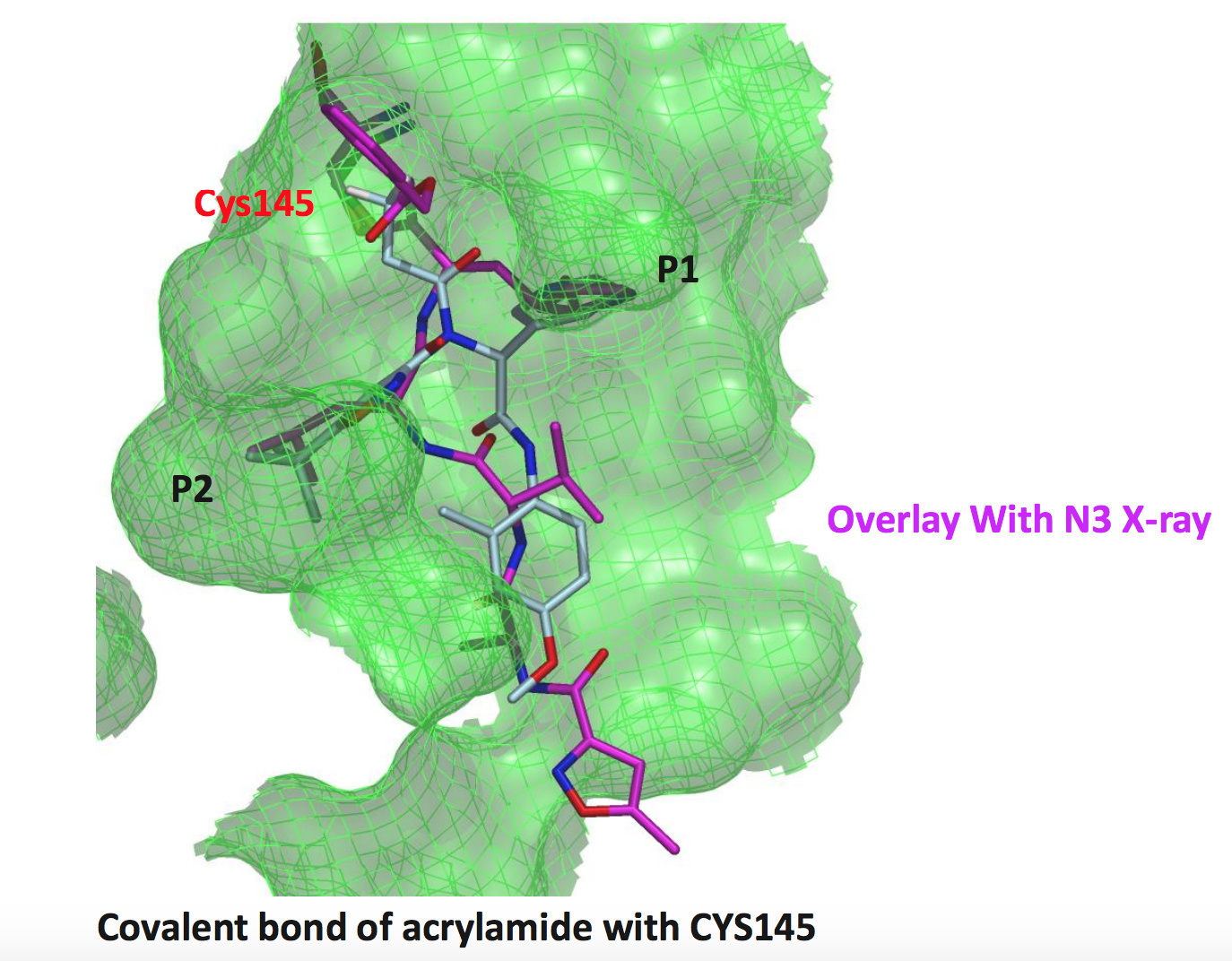
a chloroacetamide structure
LON-WEI-8f408cad-3 (x0689)
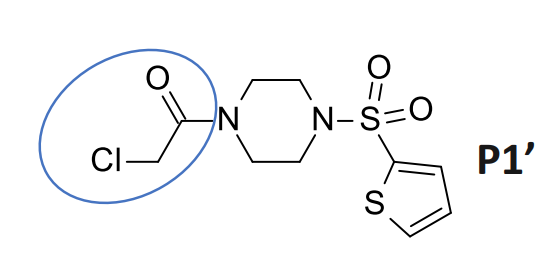
@MarkC shows that this piperazine with a chloroacetamide warhead (likely very reactive) reaches into the P1’ pocket, where the Ugi compound (also shown) does not reach.
Obviously, understanding the reactivity of both the chloroacetamide and acrylamide warheads, and how they contribute to reversible/irreversible inhibition is vital, which I’ll go into more a bit later.
A non-covalent 3-aminopyridine structure
TRY-UNI-714a760b-6 (x2646)
Fluorescence IC50: 24.57 uM
RapidFire IC50: 13.90 uM
This is one of the compounds that most of the med-chemists of the group immediately zeroed in on due to its relatively good ligand efficiency.
As you can see from the structure, the compound spans the P1-P2 sites 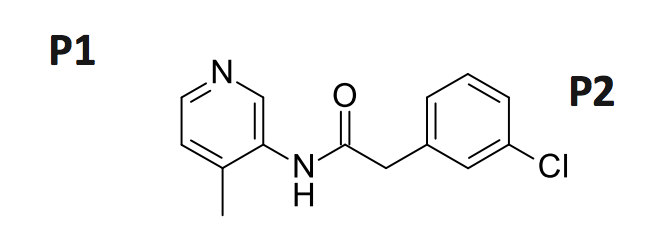
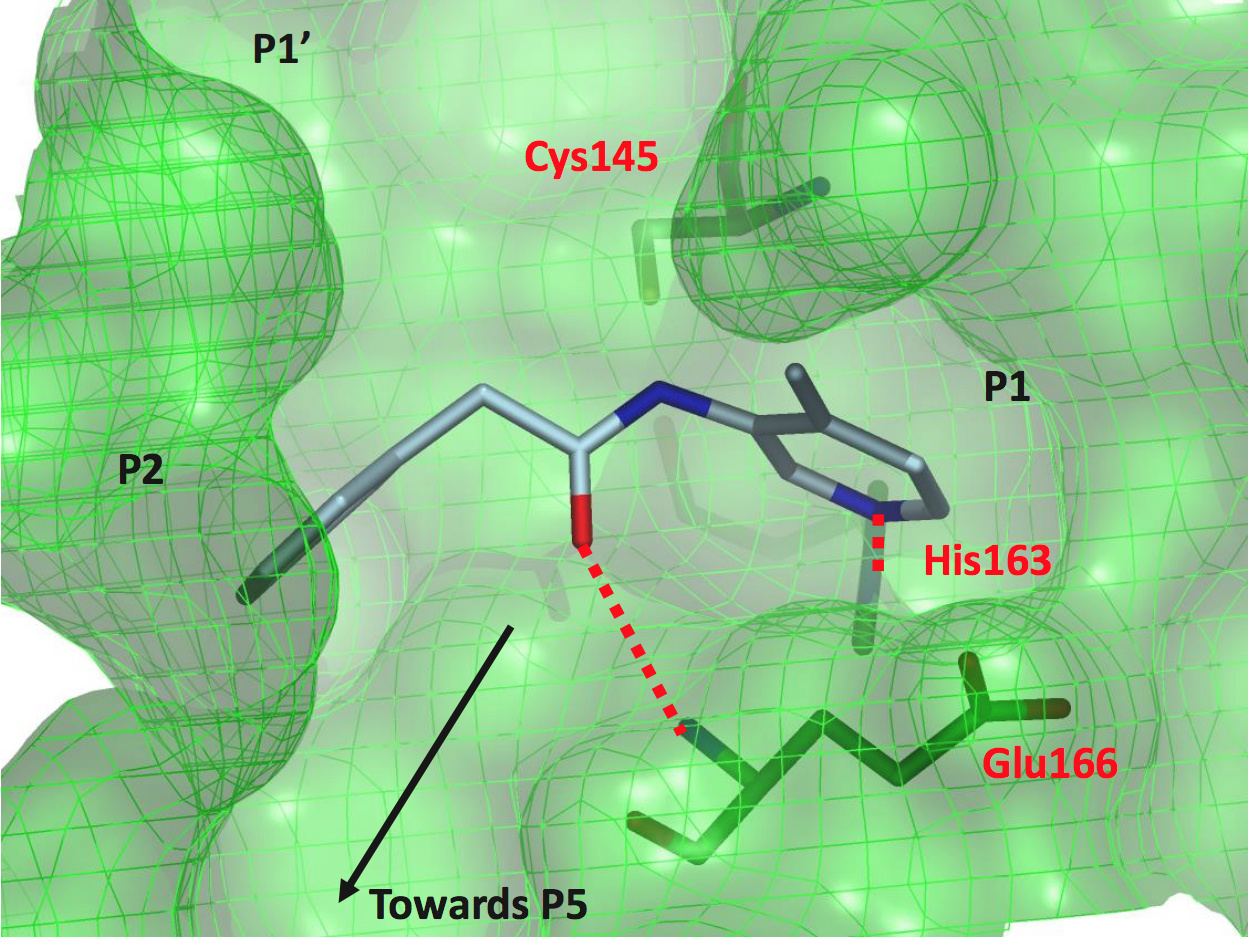
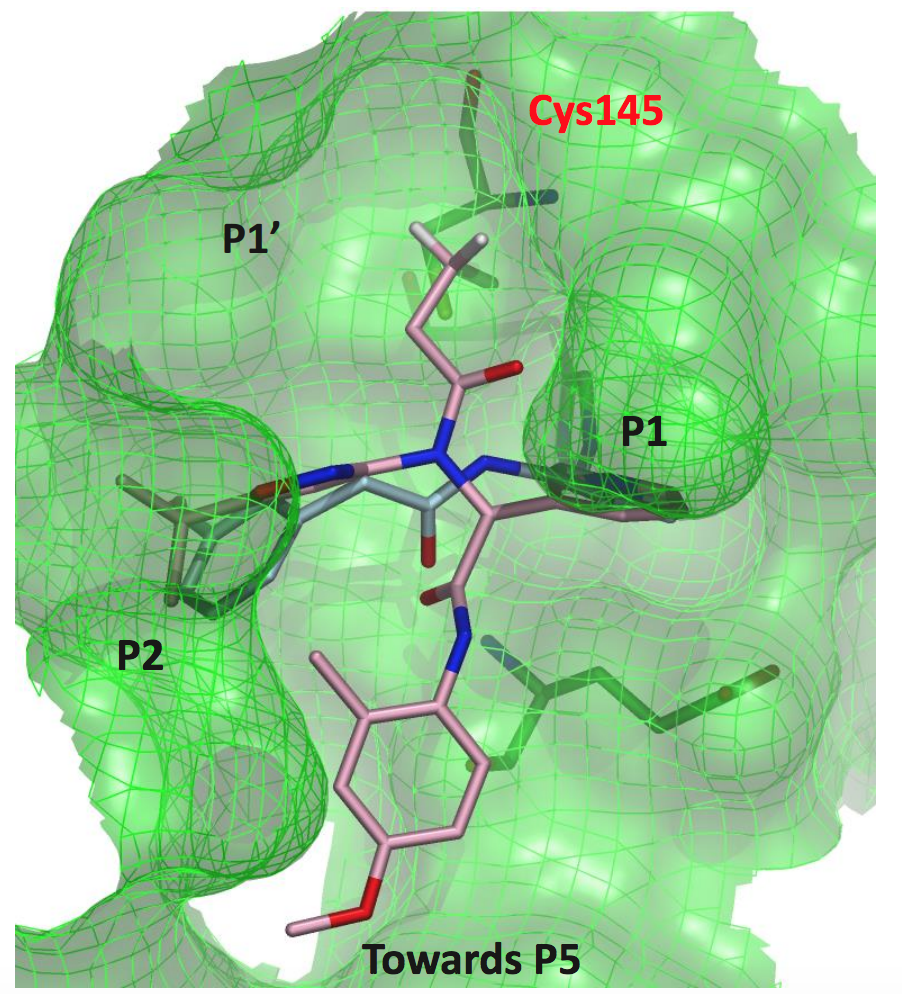
The overlay with the Ugi compound shows that there is still a lot of room to explore in orthogonal directions.
a non-covalent quinolone structure
MAT-POS-916a2c5a-2 (x2910)
This one is also non-covalent and spans the P1-P2 pockets.
| IC50 (µM) - Fluorescence: | 5.83 |
|---|---|
| IC50 (µM) - RapidFire: | 3.46 |
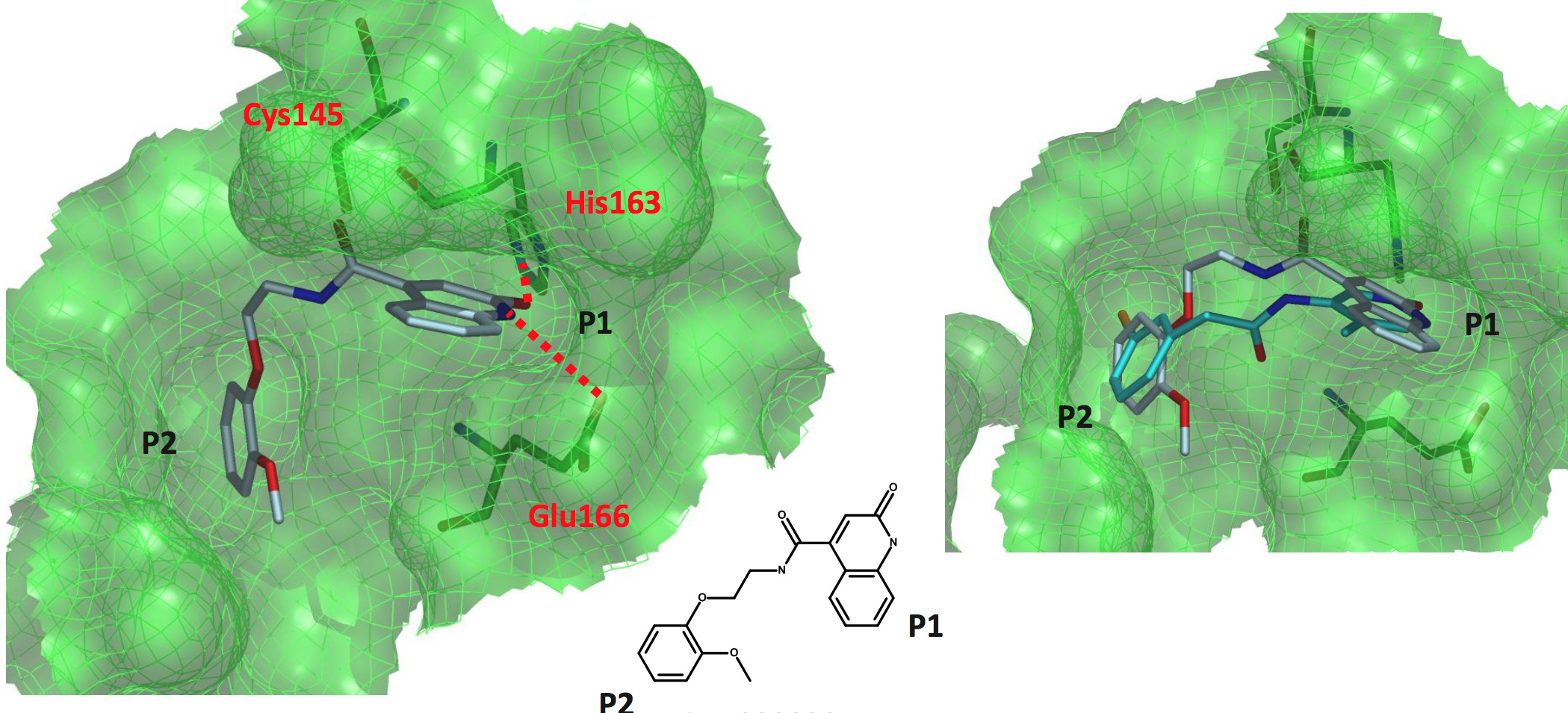
The overlay with the 3-aminopyridine above shows the clear difference in the linker binding.
I’ll also note that this one was dug up as I was reading through a 2009 high throughput screen of the original SARS-CoV main protease https://pubchem.ncbi.nlm.nih.gov/bioassay/1890 . It’s nice to know that the activity we observe is quite similar in this main protease.
In related news, @londonir has done a great job of setting up a new HTS at Weizmann for this MPro. It’s about halfway done, and some initial hits (still need confirmation) can be seen here
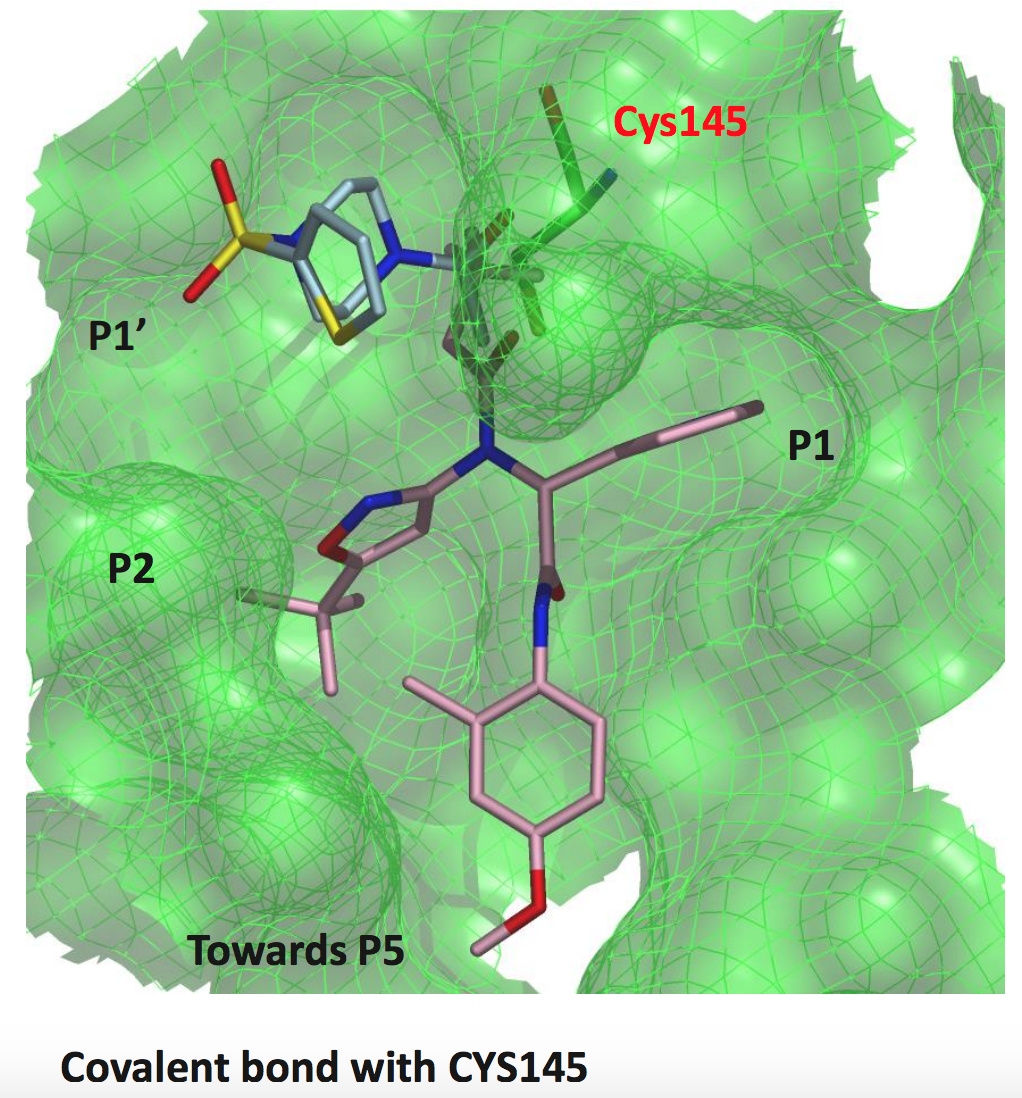
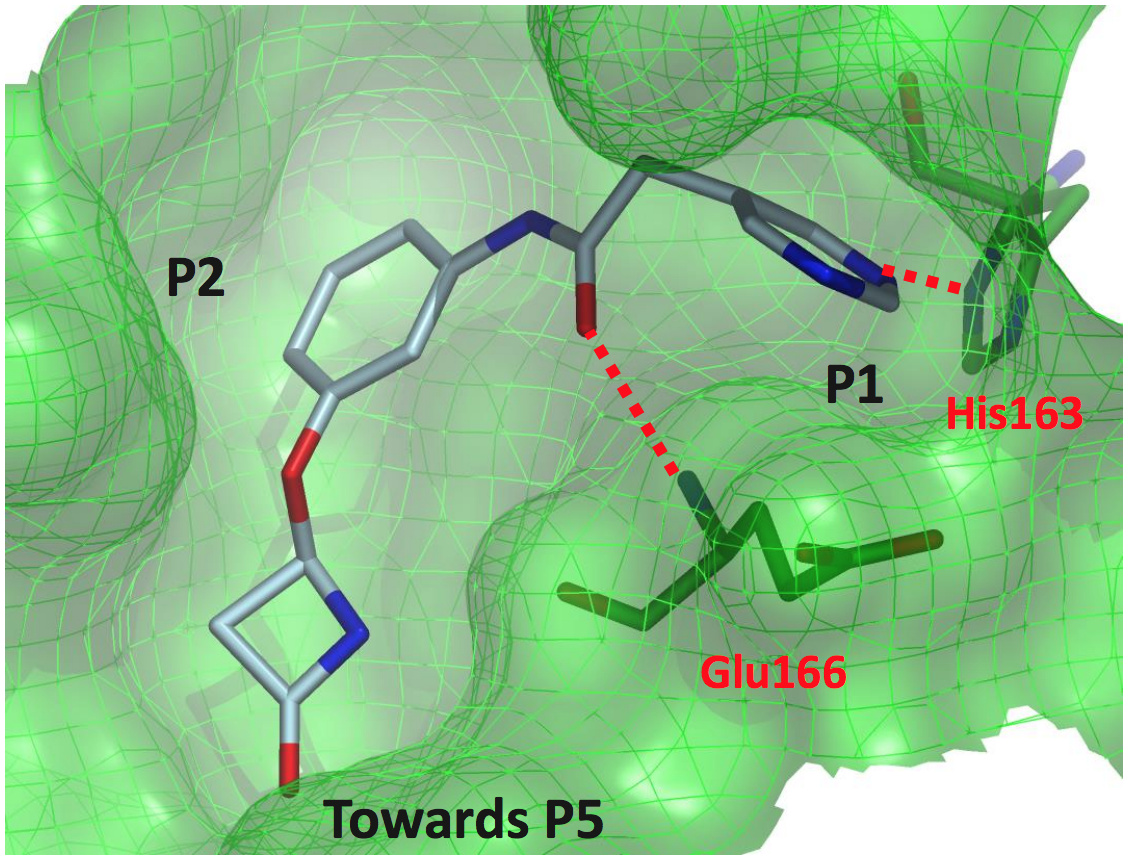
one last Beta-Lactam structure
BAR-COM-4e090d3a-39 (x2562)
This one is actually a weak binder (22.7% inhibition at 50 uM), but was of interest because the lactam binds closer to the p5 pocket.
I’ll mention that this was a whirlwind overview of the structures. It’s worth exploring them (and the many more available structures) yourself on Fragalysis. Getting into the details, you should also check out @RGlen’s excellent points here Tour of Learning from Docking, modelling and crystal structures. He knows this structure really well, and there are some nasty, intricate details that he covers.
Expanding on the Initial Hits
We’ve actually been able to very quickly do quite a lot of exploration of the 3-aminopyridine and Ugi scaffolds – just by exploiting quickly made compounds. See all the results so far at https://covid.postera.ai/covid/activity_data
@londonir’s lab has led the exploration of the Ugi scaffold. I looked through it, and we have so far assayed ~120 Ugi compounds. In order to better understand what was happening, I made a table like this of the activity data, which may be useful to some people (3 most potent compounds shown)
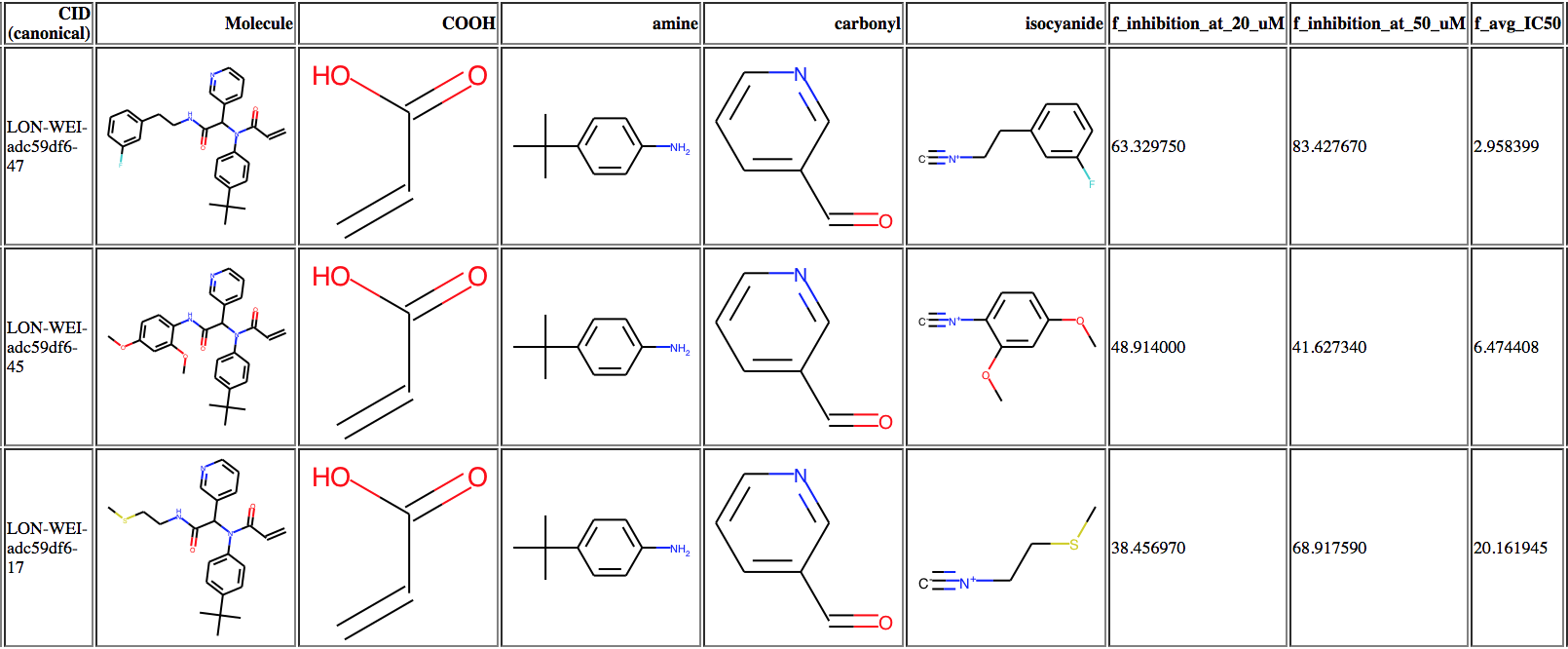
The full table is available here
You all have partially led the initial exploration of the 3-aminopyridines – and many more compounds are on the way. JOR-UNI-2fc98d0b-12 is down to 3 uM; however, we are yet to see any massive gains in potency over TRY-UNI-714a760b-6. But we have some SAR along the way!
Also, if you haven’t yet, definitely check out @pwalters blog post where he explores some of these relationships.
I have to say we didn’t do the best job of making people mention which series they were expanding upon. Therefore, I had to write some nasty SMARTS strings to grab the current expansion assay data, but here it is for the amino-pyridines if you want it! (will also be up on the site soon):
amino_pyridine_activity.xlsx (498.2 KB) (more strict 3-aminopyridine scaffold)
amino_pyridine_like.xlsx (536.7 KB) (slightly less strict scaffold)
We also have very preliminary exploration of the quinolone scaffold, which I am attaching here if you want to play around with the data yourself:
quinolinone_activity.xlsx (84.9 KB)
Lastly, there are quite a few piperazine-chloroacetamides at the top of the activity data list https://covid.postera.ai/covid/activity_data The improving potency despite a relatively constant warhead structure did hint that some improved recognition was happening. However, with these chloroacetamides understanding the reactivity is key. Which brings me to my last point… we finally have some very preliminary K_inact/K_i data, as many of you have been asking about for a while
K_inact/K_i
First, a big thank you to @Wal-Ward, who reached out to offer his enzymology expertise in analyzing some of this data. [Update: he has kindly posted his initial analysis here Biochemical assay kinetics] I’ll reiterate that these results are very preliminary.
@londonir had his group measure K_inact/K_i for 13 of the most potent covalent compounds.
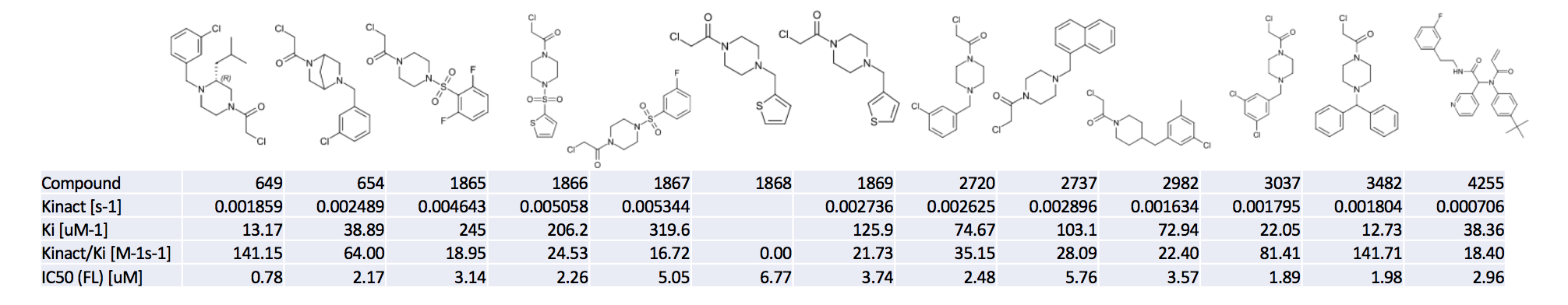
As is expected, the chloroacetamides are quite reactive (if we take K_inact as an imperfect proxy for “reactivity”). The sulfone-piperazines as especially reactive. The Ugi acrylamide has by far the lowest K_inact, as expected – though the K_i still could be greatly improved. Warheads aside, some of the med-chemists pointed out that it also looks like potency is being driven by lipophilicity in a few of these compounds.
For reference, @londonir provided the K_inact values for some approved irreversible kinase inhibitors:
5.60E-03 Acalabrutinib
2.70E-02 Ibrutinib
1.80E-03 Dacomitinib
2.40E-03 Afatinib
1.10E-03 Neratinib
1.1E-01 Osimertinib
These aren’t terribly different values from some shown here. The difference is that the K_i in those drugs is orders of magnitude better. Room for improvement.
In the next couple of days, hopefully we will have more K_inact/K_i data and also will share the full protocol.
Last thoughts
We are now constantly pushing new data through the assays and getting back new structures (the Diamond UK beamlime is back up soon); therefore, I will try to produce these data releases more frequently!
Also note, that these results mainly represent the testing of the earliest compounds that were sent off for synthesis. We should have lots more results both exploring new, more complex scaffolds, and expanding on these early hits in the near future.
Requests
If you want to buy compounds for testing / send already made compounds for testing: Please see How to send moonshot compounds for testing Some of you have already had your compounds tested!
Questions
I’m sure many of you have questions. Please reply to this thread if so, We’ll try to respond.
Thanks
And I just want to thank once again all of the incredible people involved in this project – behind all of this data is a lot of dedicated people working around the clock.
-Matt, and the rest of the PostEra team helping out on this project (@alphalee, @aaron.morris, @ajajack, @milan.cvitkovic)