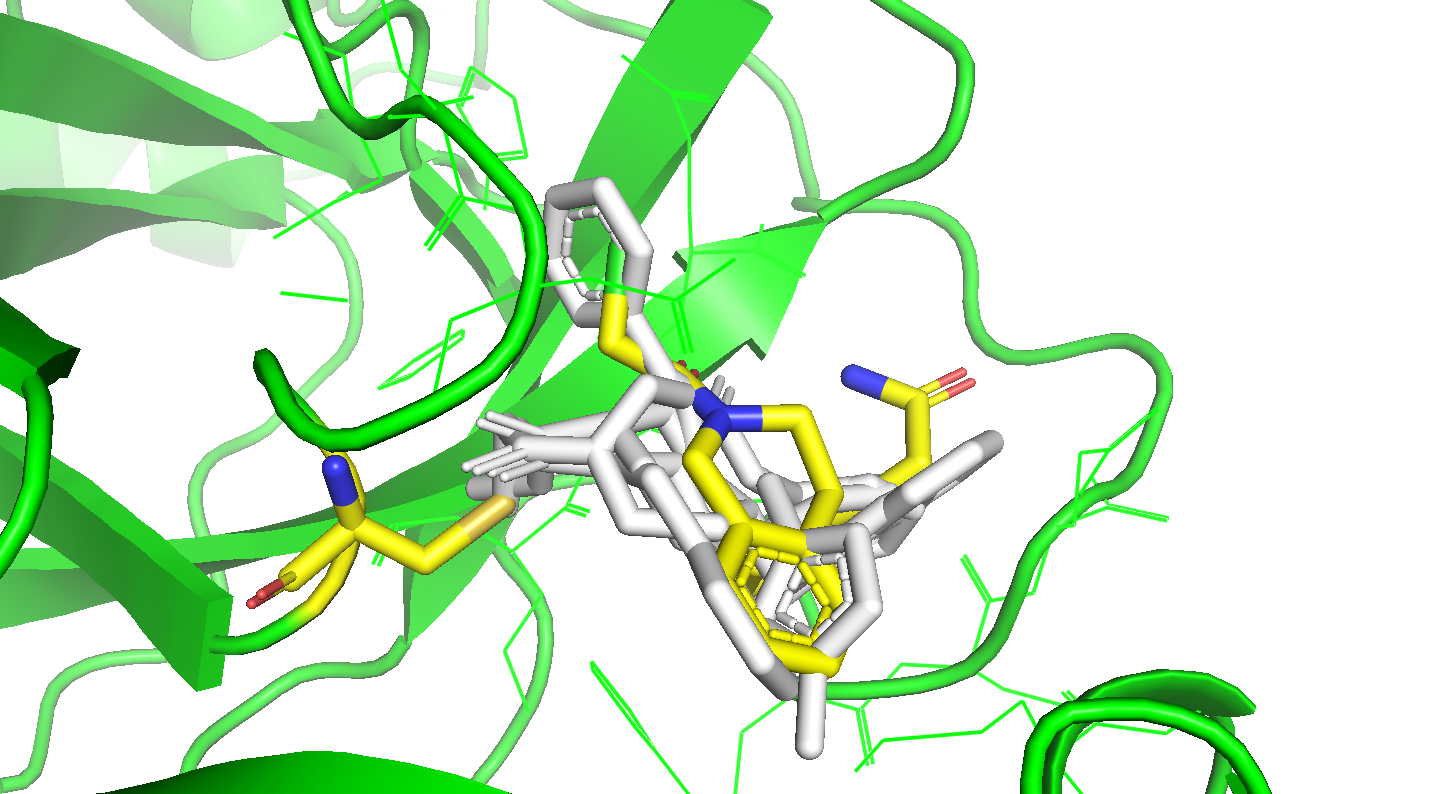
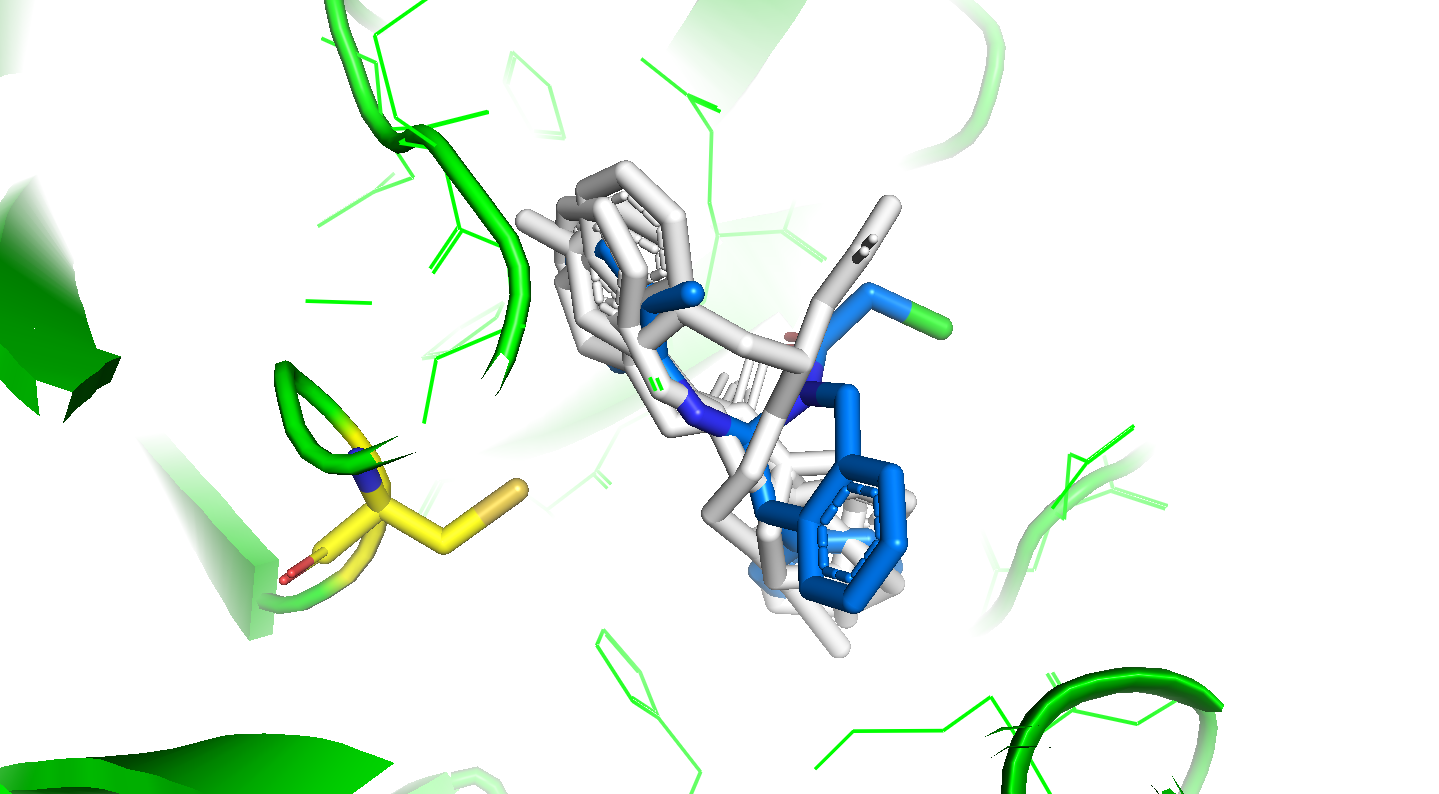
I’ve updated my Folding@home repository with constrained docking of the round two compounds containing covalent warheads with two major changes:
Compounds with covalent warheads have been docked with a constraint to be within 4A of the CYS145 SG atom
I ran an MD simulation of ~100 ps to examine the minimum and average distances of the warhead reactive heavy atom and the CYS145 SG (covalent_distance_min, covalent_distance_mean, covalent_distance_stderr)
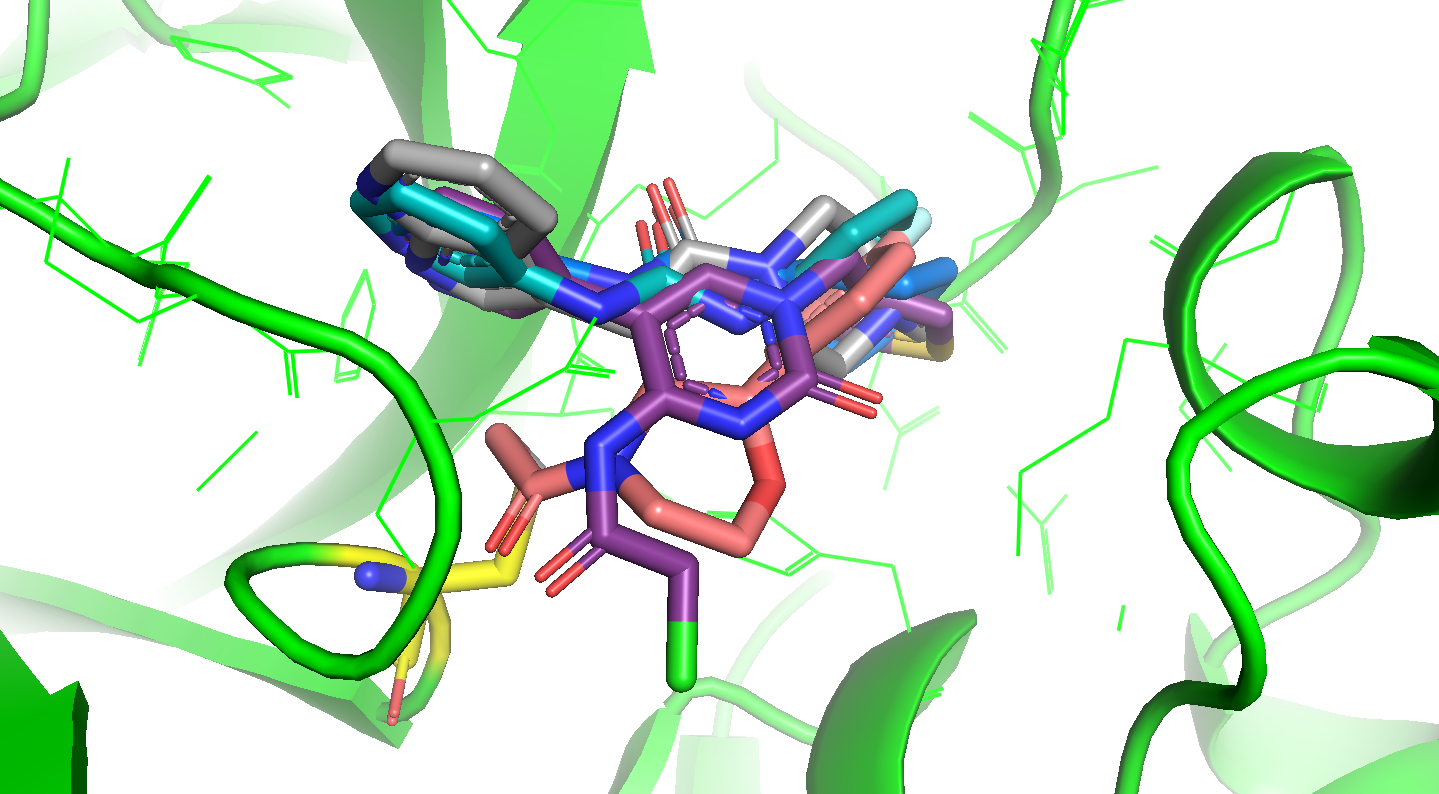
I’ve also computed an “overlap score” that assesses how well each docked pose overlaps with the shape and interactions spanned by all the X-ray fragments, and pulled out specific fragments that overlap closely with parts of the docked ligands. See the attached tarball:
covid_submissions_with_warhead_info-monomer-docked-overlap.csv - a CSV file ranked by best Chemgauss2 docking score (lowest) to worst, using a constraint to try to get the warhead within 4A of the CYS145 SG atom
fragments : the fragment listed as inspiration for the design into which the molecule docked best
Hybrid2 : the Chemgauss4 score (lower is better)
site : the type of site it binds to
covalent_distance_min,covalent_distance_mean,covalent_distance_stderr : min distance, mean distance, and stderr of mean distance (in Angstroms) of covalent warhead tagged heavy atom from @nir’s SMARTS string to CYS145 SG (should be < 4A)
number_of_overlapping_fragments : the number of closely matched fragments I found
overlapping_fragments : list of closely overlapping fragments
overlap_score : a measure of how much of the space spanned by fragments the molecule occupies
volume : the volume of the molecule (A^3)
covid_submissions_with_warhead_info-monomer-docked-overlap.sdf : same info, with structures of docked compounds
covid_submissions_with_warhead_info-monomer-docked-overlap.pdb : only structures of docked compounds
covid_submissions_with_warhead_info-monomer-docked-overlap.mol2 : only structures of docked compounds
covid_submissions_with_warhead_info-monomer-docked-overlap/ : directory of overlaid fragments for each docked molecule. The docked molecule appears first, and any fragments over a threshold appear as other molecules in the file. Not all docked molecules have highly overlapping fragments, so some of these files only contain the docked molecule.
I would use this by working from top to bottom of the list (best to worst docking score) and then eliminating ligands with poor noncovalent CYS-SG distances, then identifying those that overlap with the most fragments and have large volume occupying the binding site.