Dear PostEra organisers,
Would there be a possibility to (automatically):
- Assess similarity assessments across submissions
- And/or assign unique design identifiers to allow the identification of identical design submissions
(e.g. like a LiveDesign V number?)
Thanks for your feedback,
Chris
Hi, thanks for the question!
Are you imagining perhaps clustering (such as butina clusters) and then showing which submissions came came up with similar ideas. Or perhaps a grouping of submissions by fragments used? And seeing the diversity of submissions coming from the same fragment inspirations? Happy to try to set something up and do some analysis once we get all the first round of submissions in.
Yes, this is a known problem. Each submission is set up with an ID (such as CHR-SOS-e96). And each molecule within that has a number (1-6, in that case). However, this is not the best system. I am not aware of a LiveDesign number, is that just a unique ID?
And I checked last night, pretty amazingly, across the ~450 submitted molecules so far, there has not been a single duplicate molecule.
Many thanks mc-robinson, your proposed solutions regarding clustering and grouping based on fragments used indeed will be very useful to stimulate joint collaborative design efforts.
I noticed you are providing intermediate overviews, thanks, considering designs of other contributors will provide useful templates for cross-over and iterative refinement of ideas.
Cheers,
Chris
That is really cool! Shows people from different groups have different thinking, and that we can potentially get a much richer set than just focussing on one method/group of chemist. Still amazed that you built this useful interface in two days Matt
Thanks Matt, Anthony,
Related, would it be possible and useful to organise e.g. a short teleconference opportunity with all participants to discuss and exchange design strategies, etc.?
Such a virtual meeting can also stimulate e.g. feedback and collaboration on different approaches and pointers to experimental data/insights to help prioritise designs together?
Cheers,
Chris
This is an awesome idea Chris. Let me talk to the guys in the afternoon UK time, and perhaps we can organize a zoom meeting through Twitter! It would definitely be good for people to bounce ideas off of each other
Interesting discussion, one other possibility might be to run t-sne/umap/tmap to visualize the “chemical space”, although this requires an interface with chemical awareness (i.e. when you hover over a dot you can see the structure).
For clustering, on small/medium sized datasets usually affinity propagation works quite well for me, mostly because you do not need to define the number of clusters upfront.
If any help is need running these kind of algorithms let me know!
Thanks for the input Bart! Visualizing chemical space is a great idea, but I agree that those plots are pretty useless without the chemical awareness. (I have done this before using the very nice plotting software Altair)
I also think the main problem with tSNE, is you often change the settings to show what you want to show, e.g. https://distill.pub/2016/misread-tsne/ .
And I’ll definitely look into running the affinity propagation and let you know If I need your help!
Out of curiosity I run the AP clustering  :
:
The .pdf and .sdf are in the drive below:
https://drive.google.com/drive/folders/1FN_Bqb70VCuHvQ4ELGaUf7qqB4sibaA-
@bart.lenselink, this is super cool. Let me try to find a good way to display the data on the website. Out of curiosity, do you have any code you can share so I can update results?
Not yet. But I will try to see if I can reproduce this in rdkit+sklearn tomorrow.
1 Like
Ok, here is my first try in rdkit+sklearn:
- README still to be added (but see below)
- Currently it is still based on morgan FP only, in the Pipeline pilot +R version I’m using, physchem properties are also included.
Installation through the .yml file.
Running:
python cluster.py -i covid_submissions_all_info.txt -o covid_submissions_all_info.sd -damping 0.8 -max_iter 1000 -convergence 100
last three are AP clustering settings. Let me know if you need anything else!
1 Like
This is awesome @bart.lenselink, thanks! I will try to pull something together tonight.
1 Like
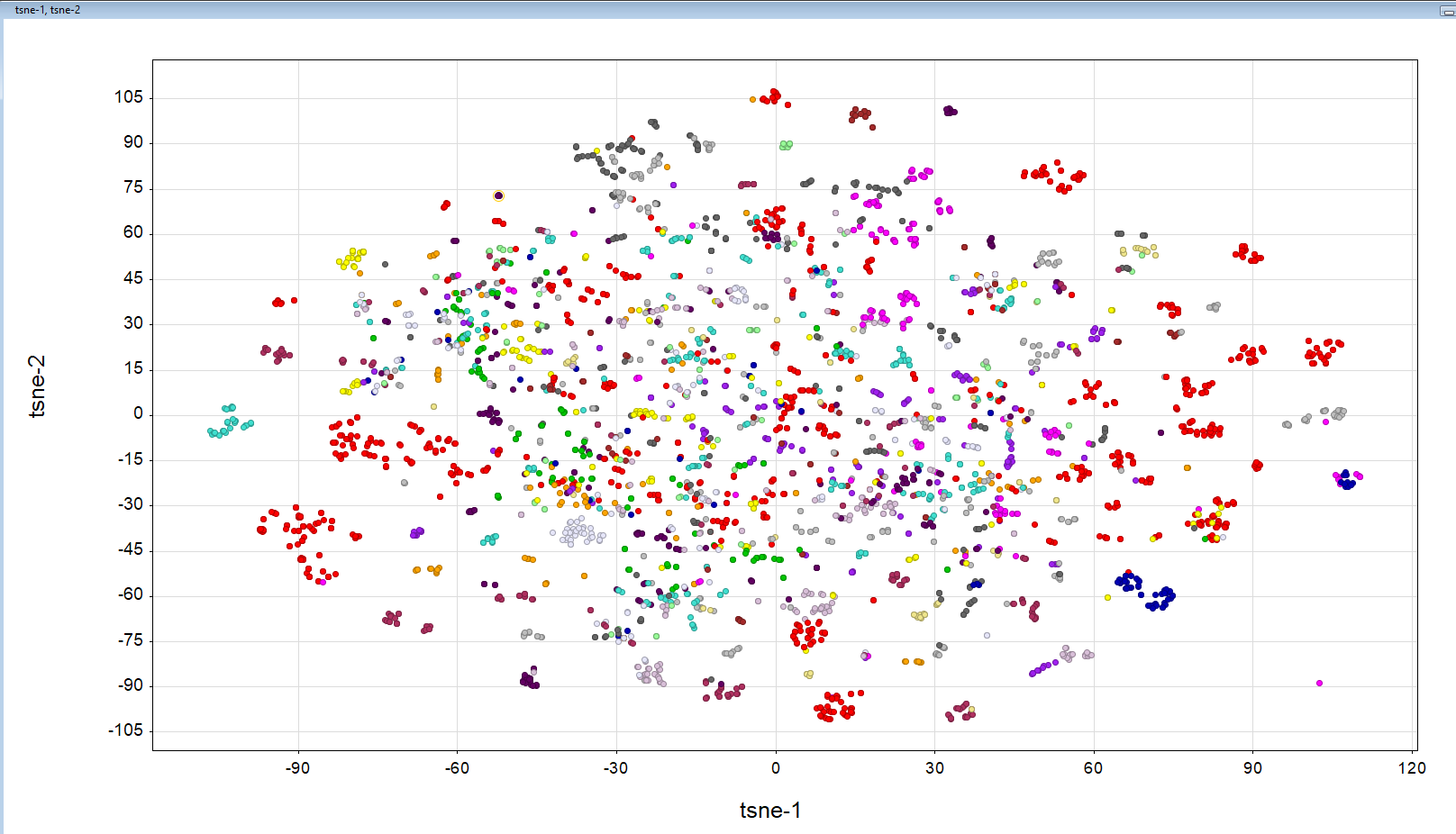
By the way, we pulled together a brief PCA plot to see if compounds clustered at all. Pretty much one big blob… Perhaps time to mess around with some t-SNE parameters until a pretty picture emerges 

I’m also still working on getting cluster results such as yours up in a viewable format!
Nice- regarding the PCA- the same is the case with t-sne it seems, perhaps the perplexity could be adjusted to get more clustering. But at the same time, after hovering over the clusters that are well separated you can see the clusters do make sense. In this case it is colored on the submitter.
Reproducing this should be easy- just add/replace the following:
tsne = TSNE(perplexity=float(args.perplexity), n_components=2,init='pca').fit_transform(FP)
df['tsne-1'] = tsne[:,0]
df['tsne-2'] = tsne[:,1]
PandasTools.WriteSDF(df, args.o, molColName='Molecule', idName="CID", properties=list(df.columns))
In this case perplexity 10 was used.
1 Like
@bart.lenselink So finally got to playing around with this a bit, initial results are here: https://github.com/mc-robinson/covid-submissions-viz
Annoyingly, Github does not render the bokeh plots correctly so I need to actually get it up on the site to be seen. However, you can clone the repo if you wish to see the interactive bokeh code (graciously adapted from here: https://www.macinchem.org/reviews/fdamols/interaction.html). I’m currently using Molecular Weight as the coloring, but I think using the creator of the submission may be more informative. Will keep playing around with it!
1 Like
What’s needed during the submisssion process is to perform a quick check of the most similar molecules - including fragments screened and submitted designs - so that users can see if their suggestion is novel, what is known about similar molecules, or if there are errors.
I guess a quick pullback to display the 10 most similar should be sufficient - allowing users to go either cancel, modify or continue with their submission.
2 Likes
@krisbirchall. We are working on developing a similarity search portal – so that could be quite useful for what you are proposing. Currently, we only calculate similar molecules once the molecule has been submitted, but your proposal does make some sense, so let me think about the implementation details.
My understanding of chemoinformatics is rudimentary, so my question might seem a little simple for experts.
How is this clustering of these “similar molecules” conducted? There are quite a number of measures I can think of, e.g. shape screens, Tanimoto similarity, log P (or ClogP), polar surface area (PSA), and other physicochemical properties.
So when the clustering is conducted, are each of these factors assigned a weight and scored accordingly? Or in the case of @mc-robinson 's similarity search portal–would it likely take these factors into account?
Hi @Zhang-He – the current approach taken on the search portal is indeed to use Tanimoto similarity based off the Morgan-3 fingerprint representation of the compound. So currently the similarity doesn’t directly use the physicochemical properties you mentioned.
1 Like