Summary
Drug discovery is hard. Artificial intelligence and Machine Learning approaches are being deployed widely to help make it easier. But many chemists remain skeptical given the past hype that has failed to deliver and the mess that is the ML literature in other fields recently highlighted by Derek Lowe and others.
But some of us working in the field do attempt to think seriously about proper validation and understand the limits of our work. One natural question is to ask what work would be left even if current ML/AI approaches get to near perfect performance? And what types of problems remain beyond the reach of the machines? A new report of a rare disease small-molecule discovery campaign provides a partial answer.
A Truly Small Molecule
Amid the many impressive articles in a recent issue of J. Med. Chem reporting novel scaffolds, macrocyclic linkers, prodrugs, and even binders found through a combination of DEL screening and PROTAC synthesis, was a curious paper impressive for the simplicity rather than the novelty of its chemical structures.
This is not at all to demean the paper, Identification of 2,2-Dimethylbutanoic Acid (HST5040), a Clinical Development Candidate for the Treatment of Propionic Acidemia and Methylmalonic Acidemia which told of the excellent efforts by the team at HemoShear Therapeutics to find a clinical candidate for two extremely rare metabolic diseases. In fact, the non-paradigmatic drug discovery campaign – with a non-protein target, hardly any chemical synthesis, and seemingly no hit expansion or lead optimization phases – brings into focus precisely those areas of drug discovery that AI systems do not fully address yet.

An image encapsulating the wide diversity of drug discovery campaigns. The top paper is the inspiration for this post, while the bottom paper combines the recent innovations of DEL and PROTACS to arrive at a novel degrader. The clinical candidate identified in the top paper has only 8 heavy atoms while the degrader discovered in the bottom paper has 81 heavy atoms! From https://pubs.acs.org/toc/jmcmar/64/8
What makes this paper so unique
The word “Identification” in the title of the paper gives a hint as to the special nature of the project. The clinical candidate, 2,2-Dimethylbutanoic acid is available off the shelf for testing. In fact, as far as I can tell, every molecule tested for the project is readily available off the shelf, as the https://postera.ai/manifold screenshot shows below.
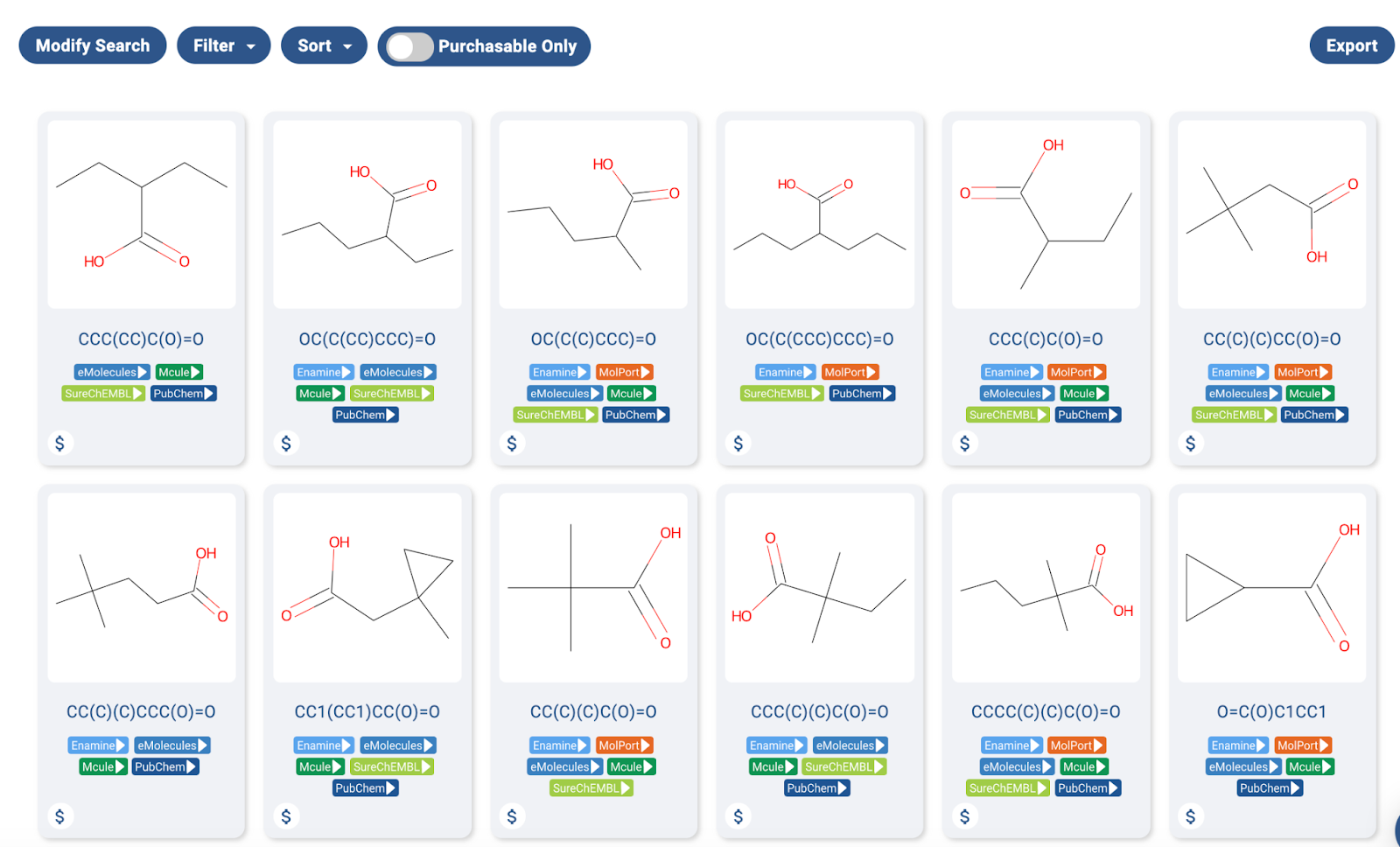
A screenshot of a Manifold search of some of the acids used in the project. As one can see, the molecules are readily available in-stock from multiple sources.
One may wonder why every molecule tested is essentially a small carboxylic acid, and this is where the group’s deep expertise in the disease process was essential. I’ll try my best to describe the group’s strategy here, but I am decidedly not an expert in metabolic disorders.
To quote the paper, “Propionic acidemia ¶ and methylmalonic acidemia (MMA) are rare autosomal recessive disorders of propionyl-CoA (P-CoA) catabolism” The disorder results from lacking one of two enzymes in the pathway to break down P-CoA. And since one of these enzymes is lacking, there is a buildup of P-CoA, and other metabolites inside the mitochondria that can disrupt other metabolic processes. Unfortunately, this often means morbidity and mortality in youth, and organ dysfunction or failure for older survivors. It’s not a nice disease.
Using their in-depth disease knowledge, the group hypothesized that “low-molecular-weight carboxylic acids that can readily form CoA esters using native human cellular enzymes could lead to a redistribution of the cellular acyl-CoA pools.” Thus, they ordered ~30 carboxylic acids and tested them. One of them worked, and is now in clinical trials!
Essentially, one can imagine the one round of molecular design work done on this project as a few substructure search and filtering steps, as shown below:
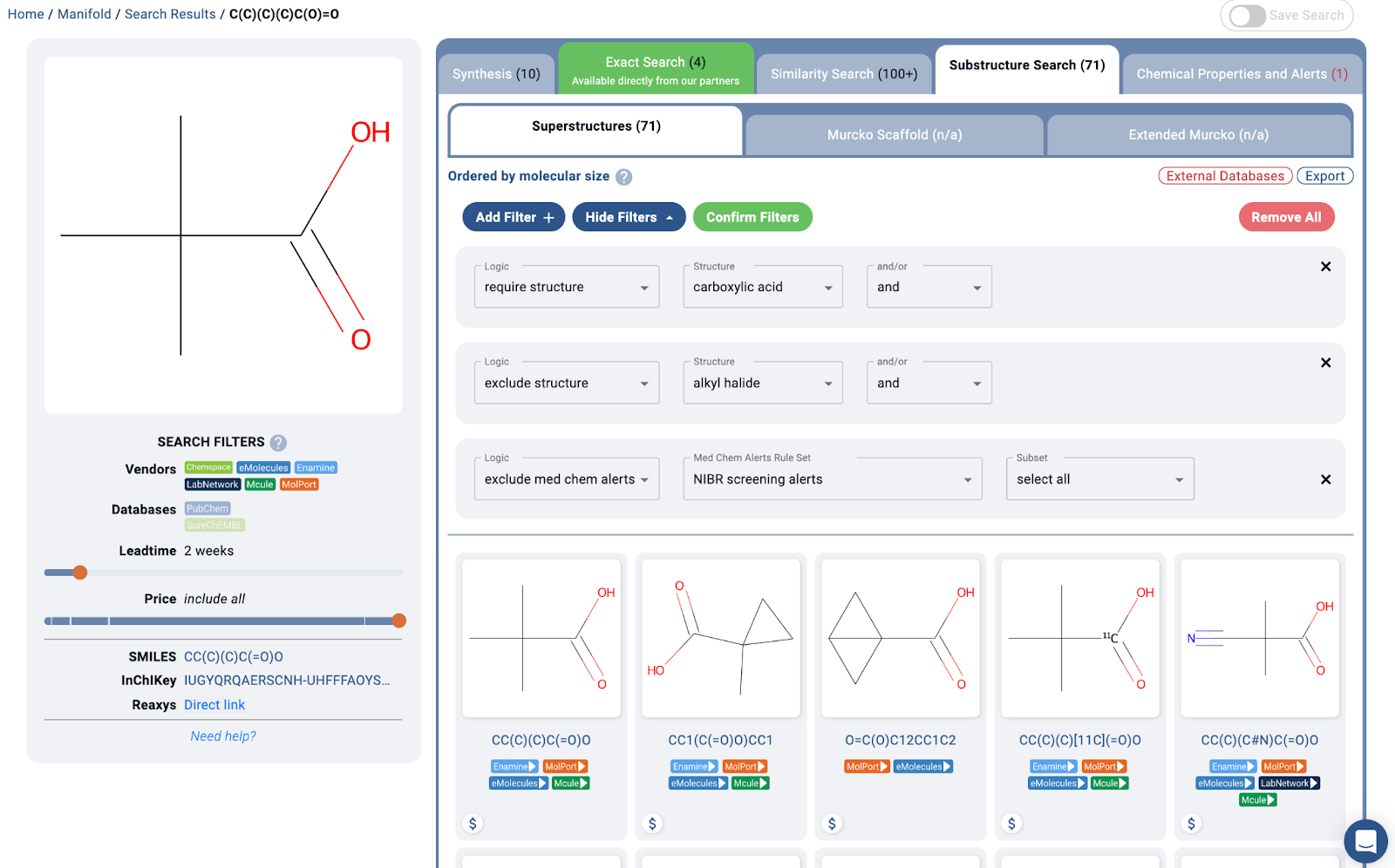
A https://postera.ai/manifold search showing a simple example of how molecules may have been found for the project. Of course some extra medicinal chemistry expertise was added in preferring structures such as the quaternary alpha carbon, thus avoiding some problematic oxidative metabolism pathways.
Let’s recap what makes this project so unique besides the extremely rare disease area:
-
The target is not a protein, but rather the redistribution of an accumulated small molecule.
-
No follow-up hit to lead and lead optimization campaigns. This was essentially one-shot drug discovery built on a very specific, and seemingly successful hypothesis.
-
The extent of chemical synthesis undertaken for this project was the single cGMP synthesis of the clinical candidate shown below.
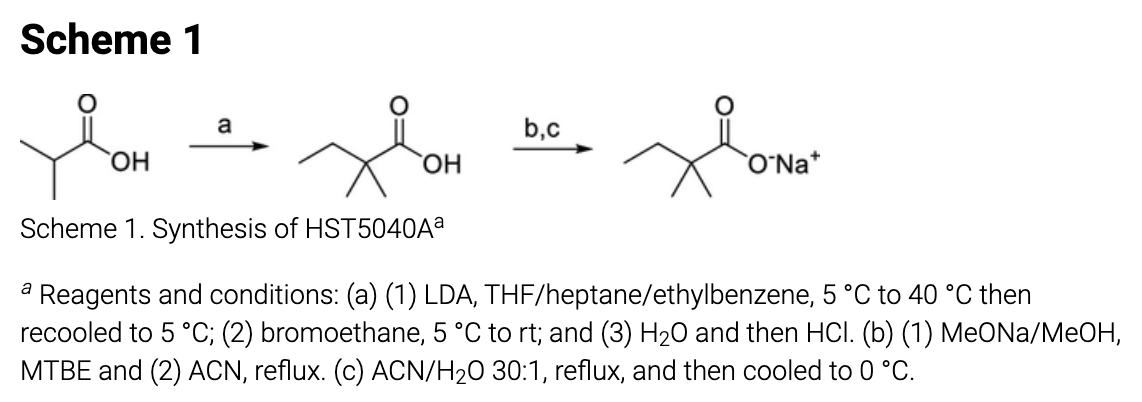
The single synthetic route included in the paper. From https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00124
Thus, there was ostensibly no need to design a second round of analogs, and there was no prior inhibition data that could be used for training machine learning models before purchasing the initial set of molecules. There was also no structure to allow for the newest molecular simulation methods. The target itself wasn’t even an enzyme/protein, and may likely elude many of the machine learning methods being developed for target discovery and validation.
Essentially, it is a project totally devoid of the usual pains of drug discovery that ML and AI try to target 1, so how were they able to write up a whole paper? Turns out there was plenty to talk about…
All of the rest of the work
Reading the paper is basically a great case study of all of the hard work that goes into drug discovery besides the iterative cycles of designing and making chemicals. I don’t dare try to summarize all of the details, but here is some of the hard work that had to happen to get this molecule into the clinic.
-
A specialized phenotypic assay utilizing primary hepatocytes derived from actual PA patient livers.
-
Further assays to determine that the mechanism of action for the acids was indeed the desired mechanism.
-
Full characterization of the pharmacology of the lead molecule in a proprietary disease model where key biomarker data could be collected.
-
The compound was then tested in hepatocytes from humans, mice, rats, beagle dogs, minipigs, and cynomolgus monkeys. And of course there were interspecies differences… It was then determined that minipig would be the best model.
-
Extensive in vitro and in vivo ADMET profiling of the compounds, including in vivo and in vitro metabolite ID studies. Note that much of this had to be done in minipigs, due to them being the most pharmacologically similar to humans for this disease model.
Lastly, let’s note that all of this was done despite 2,2-Dimethylbutanoic Acid, the exact clinical candidate, having already been studied in Phase I and Phase II human clinical trials, with good tolerability, for another unrelated indication! It’s quite hard to imagine a molecule which one could take more quickly to the clinic.
And then of course there are still the trials, which will take time. 2
The point of all of this is not that drug discovery without design-make-test cycles is still too hard for anyone to care about AI, because dealing with minipigs and expensive human clinical trials are extreme pains. The point is for us to understand how far and how fast the current innovations in machine learning – with the focus mainly on molecular discovery, design, and optimization – can take us. Furthermore, by understanding the limits, we can aim to understand how one would improve the areas outside the current focus of artificial intelligence innovation.
Why would a Machine Learning researcher write this?
Perhaps some of you are wondering why machine learning researchers would go into great depth regarding the limits of their own field. Well we at PostEra still believe that what we work on is vital, we just don’t think it’s going to “solve” all stages of drug discovery and projects that relate to every disease.
Drug Discovery is long and multi-faceted, thus not a single technology is going to “solve” it. Not AlphaFold, not quantum computing, and not robots doing synthesis. However, gains do compound, and given the importance of the field, huge improvements in the speed and success at individual stages of the endeavor do translate into great wins for patients.
When talking to drug discovery chemists, many are so influenced by past failures that they simply say something resembling, “but the real problems with drug discovery are [whatever reasons their past projects failed].” And we would agree that imperfect knowledge of disease biology and possible human toxicities in later stage trials is a fundamental issue. But if one could halve the amount of time in preclinical hit expansion and optimization work to find these issues in trials, that too would be groundbreaking. Furthermore, for those drugs that do succeed, getting to market years earlier would indeed be a win for many patients across the world.
And generally, at PostEra, we try to work on projects where we believe our technology can have the greatest impact. Read most patents and you will see hundreds or thousands of molecule designs and syntheses. All that takes a lot of time and effort, usually multiple years. We obviously read about other projects and try to stay at the forefront of knowledge with respect to other technologies, but our innovations are more suited to the majority of drug discovery projects, rather than the exception.
We still greatly appreciate that others are working on finding off-the-shelf acids to combat ultra-rare metabolic disorders, or even looking for anti-cancer compounds that already exist in the aerospace and defense industries, it is just probably not where we will focus our attention.
None of this should be too controversial; knowing the limits of technology is important for machine learning in other fields too. Without understanding the problem and the limits of data, you basically end up with junk, like hundreds of papers on COVID lung scans that are pretty much worthless.
And lastly, though we work heavily with machine learning, our main goal at PostEra is to bring innovations to patients. They come first, not the technology we use or how cool it sounds. And to use these technologies correctly means understanding what they can and cannot do. It also means learning about the other parts of drug discovery – even the parts currently outside the grasp of the machine.
Footnotes
1 A brief caveat that one could likely imagine a way to set up this project for better interplay with machine learning techniques. If the patient hepatocytes could be used to set up a phenotypic assay for screening, then more typical hit to lead drug discovery may have been possible. However, it is unclear if small carboxylic acids would even be included in the screening deck, and the project would have included many more compounds.
Furthermore, ML could be used to help give alerts to likely toxicity and metabolism issues, such as beta oxidation pathways of the acids without quaternary alpha carbons. So there are still significant places for ML to play a role here.
2 For some insight into how fast they can go, Derek Lowe gives the nice example of Molnupiravir for COVID, which is still not through PH III trials.
here’s a known compound, already investigated against a number of viruses (in vitro and in animal models), with a promising mechanism of action that gave some confidence that it would have activity against a new RNA virus pathogen, and which was already heading towards human trials. You could not ask for a better-positioned small molecule; this is as good as it gets. So the time it’s taken to get the clinical read on whether it really works or not is about as fast a development as you’re going to see. Everything was lined up perfectly and a lot of the work had already been done. Keep that timeline in mind as a baseline!