

I downloaded those papers about 6 months ago, it was about time I read them.

Yes adding them on the list!

DAR-DIA-5ff57136-19
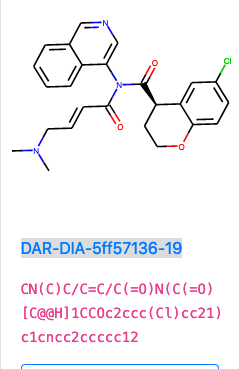
I like this one, it adds hydrophillicity via the protonated amine and should be more soluble, as well as the warhead being a soft reactant.
Bobby

It might be an idea to use the 3-chlorobenzyl P2 substituent to explore warhead variations rather than dihydrobenzopyran (or other bicyclic variations). This avoids complications associated with chiral centers and the more rigid bicyclic P2 substituents may even compromise covalent bond formation. Potency gains from appropriate rigidification of reversible inhibitors such as ADA-UCB-6c2cb422-1 are modest (< 5-fold) and I’d anticipate that inappropriate rigidification of molecular structures will result in substantial reductions in activity (whether the inhibition is reversible or irreversible). Attaching sp3 carbon to the amide nitrogen (designs 13 – 17 of the submission) would be expected to flip the conformational preference of the 3-pyridine amide from trans to cis and the effects of this can be seen by comparing IC50 values for ADA-UCB-6c2cb422-1 and its N-methyl analog MAT-POS-bb423b95-7).
2 replies
Hi Bobby,
While incorporation of a basic center in an inhibitor structure would be expected to increase aqueous solubility under physiological conditions, it is also likely to concentrate the inhibitor in lysosomes and other acidic compartments within cells. Merck Frosst researchers (who discovered the cathepsin K inhibitor odanacatib) have expressed a view [ 1 | 2 | 3 ] that basic centers in cathepsin K inhibitors are undesirable and I’ll mention @bayly in case he would like to add anything.
Both good points. I’ll submit the chlorobenzyl analogues now.
I assume that the mPRO acts, as the dimer, after transcription and assembly in the ER to perform its protease activity. mPRO is very pH dependent for its activity. It works best at pH 7.2. So, maybe we can assume that is the pH of the compartment it evolved to work in. However, I get your point we could lose compound to other vesicles. The endoplasmic reticulum (ER) is nearly neutral, the Golgi is mildly acidic and the secretory granules are more acidic (pH 5). Other compartments are definitely acidic. That’s how some of the anti-malarials work, by altering pH in these vesicles. The more acidic the vesicle, the less chance of escaping that environment for a protonated species through a membrane I expect; caveat - given passive diffusion. There are some protonated protease inhibitors e.g. indinavir, nelfinavir (in Hep C, this seems a comparable system in terms of virus assembly), although not as many of the compounds in the clinic are protonated as one would expect from a random sample, I believe that there is definitely a propensity for cell active compounds of this type not to have ionisable groups, but not exclusively. Probably as they enter the cell mostly through diffusion, although there are a number of HiV protease inhibitors that are MDR1 substrates, and presumably other transporters, and this will also be the case for our inhibitors. So yes, not ideal to have a protonated amine, but not exclusively bad for protease inhibitors. The current challenge is really bad solubility, so we will have to explore any other tricks to increase solubility!
1 reply

I don’t think compounds like these are stable, however difluoro- or trifluoroethyl substituent(s) can be used (if these aren’t too bulky). Fluoroethylamines can be cleaved oxidatively eventually forming fluoroacetate which is toxic and a thing to avoid. Morpholine is one other option for pKa tuning. Maybe it’s an opportunity to fill P1’ pocket as well?
2 replies
F-morpholines maybe?
The presence of a basic center becomes less of a concern for inhibitors that don’t hit anti-targets in the acidic compartments. My guess is that the risk of hitting another cysteine protease will be greater for covalent inhibitors and I generally assume that assessment of selectivity for irreversible inhibitors is more challenging than for reversible inhibitors.
I believe that the potency advantage of benzo-fused rings over unfused rings at P1 may depend on substructural context and I’ll point you to a discussion that may be relevant. Aza substitution of the isoquinoline ring typically leads to reduced potency but increases in aqueous solubility might compensate. Here are designs from @Ben_DNDi (BEN-DND-4f474d93) and @mc-robinson (MAT-POS-f7918075) that feature naphthyridines. This 2009 article presents analysis of the effects of aza-substitution on physicochemical properties and @edgriffen may have more current data. As discussed in the article, N-methylation of acyclic amides typically results in increased aqueous solubility. It actually works particularly well for anilides but it also inverts the cis/trans geometric preference of the amides which will result in loss of potency.
One way of incorporating a charged group would be to use an acid rather than a base, perhaps with an acidic heterocycle at P1. My guess is that S1 site would not be able to compensate an anionic substituent for its lost solvation although I’d be very happy to be proven wrong on that point. I’ll mention @JohnChodera since there could be a role for free energy calculations if going in this direction.
The worries about bases don’t go away once pKa goes below 7. My understanding is that lysosomal pH is 4-5 and pH in some compartments (e.g. osteoclast pits) can be even lower. It’s certainly a factor that one needs to take account of when designing inhibitors to function in acidic compartments. Acidic groups will tend to draw inhibitors out of acidic compartments and this points to a tactic that we might use is we find that we’re hitting lysosomal anti-targets.

@pwkenny, can you point to explanations for what drives the conformational preferences of amides? I’ve always assumed that it’s a steric thing, and so depends on the nature of the full subtituents, not just the first atoms.
To be clear, your rationalisation of that 50x drop in potency (ADA-UCB-6c2cb422-1 0.7uM, vs its N-methyl analog MAT-POS-bb423b95-7 43uM) sounds right - but that’s a single methyl group, rather than a whole covalent warhead.
MAT-POS-bb423b95-7 is definitely trans (see snapshot of impending structure) but obviously there is a penalty for adopting that conformation:
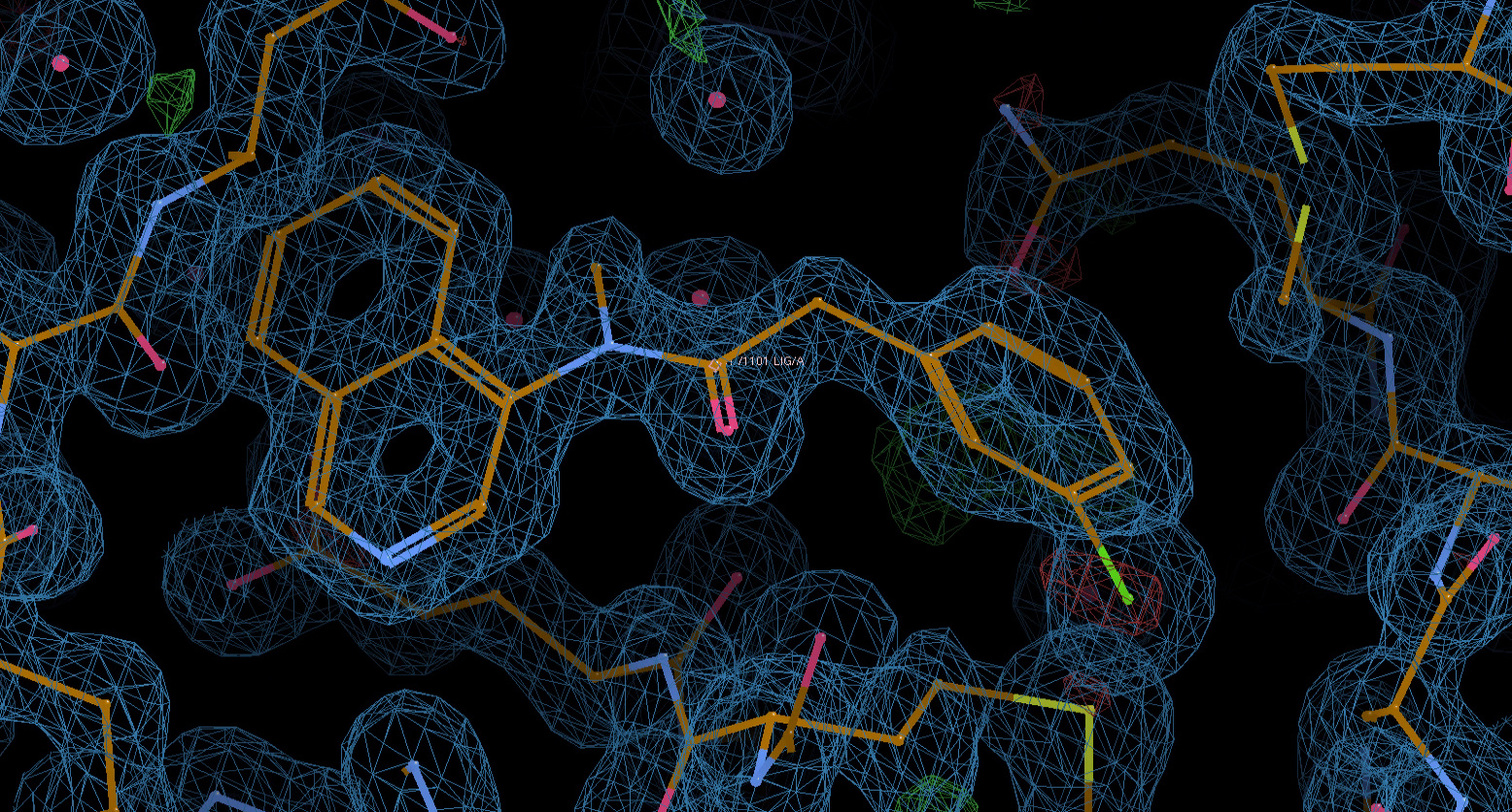
@frankvondelft some other examples of N-methylation inducing cis conformation:
https://www.nature.com/articles/srep09950
The question is whether it’s predictable what happens if it’s not just a methyl on the amine, but a methylene (or hydrazine) with other stuff attached beyond; or indeed if the carbon on the other side is chiral, rather than the methylene in MAT-POS-bb423b95-7.
I would have thought once that happens, the conformational landscape becomes complicated and must be computed / can be estimated.
1 reply
Yes - as we stick different/bigger things on either end the equilibrium will shift and could be further complicated by charge:
https://onlinelibrary.wiley.com/doi/pdf/10.1002/anie.201502474#:~:text=Abstract%3A%20Tertiary%20amides%2C%20which%20usually,strongly%20rotamer-dependent%20radical%20cyclization.
Hi Frank,
I haven’t got explanations for amide conformational preferences that don’t involve a degree of arm waving although I sometimes think of torsional preferences in terms of interactions between bonds. My view is that ‘prototypical’ torsional profiles are primarily determined by atom types (some empirical force fields are based on this assumption). If thinking in terms of substituent sterics, it can be helpful to make a distinction between the size of a substituent that is ‘seen’ by the bond and the total size of the substituent. For example, a benzyl substituent can look more like methyl than t-butyl when viewed from the bond that links it to the rest of the molecule. All that said, one can certainly use steric repulsion to perturb ‘natural’ torsional profiles (e.g. flanking ortho substituents on phenyl) and it’s also possible to engineer some gross distortions of molecular geometry using bulky groups.
One can ‘explain’ the observed trans preference of secondary amides in steric terms by observing that an oxygen atom is less bulky than a carbon atom and so it is better to have the N-Me of N-methylacetamide eclipse C=O than C-Me. However, explaining the conformational preference of N-methylacetanilide is more difficult because the less bulky substituent (methyl) is syn to the carbonyl oxygen. One way of looking at this in steric terms is to note that a methyl group is bulkier than sp2 carbon. In addition, N-methylation would push the phenyl out of co-planarity with the amide leading to increased repulsion between the pi-cloud of phenyl and the carbonyl oxygen.
Regardless of how we explain them, published experimental data (and our own IC50 measurements) are telling us that an anilide NH is not such a great vector for structural elaboration (even though the ease of synthesis algorithms may well suggest otherwise).
Hi Daren,
The data in the NMR study show that the cis/trans preference of acetanilides can be modulated by tuning the electronics of the benzene ring (although not to the extent that the conformation with benzene syn to carbonyl oxygen predominates). While molecules can be forced into ‘unnatural’ conformations, this needs to be done in a manner that doesn’t compromise affinity.
