Receptor prep and docking model optimization
This work started with the 6LU7 complex structure, since I thought the conformation of the N3-bound structure would best accommodate growing the fragments from the Diamond screening hits. I began by modeling the 6LU7 non-covalent encounter complex using the deprotonated thiolate form of C145, which forms a good h-bond with the amide NH of the N3 ligand. I ran a 500 ns trajectory of the dimer with ligand bound in one site only, using Desmond MD with SPC waters. From this trajectory, I saved 5000 frames, and generated OEDocking/FRED grids for every frame (as well as for the xtal structure model), and docked the entire dsi_poised library (768 compounds) using the OpenEye FRED program.
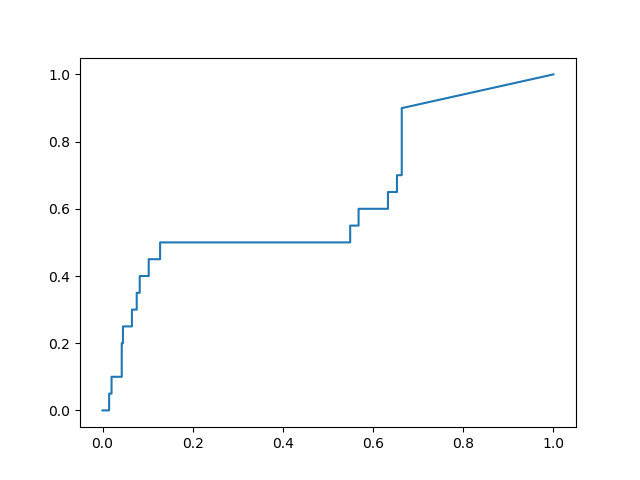
I calculated the ROC curves for recovering the non-covalent actives in the dsi_poised set published on the Diamond website. I computed the early enrichment factor for the top 100 ranked compounds, and found 3 frames that stood out as providing significant improvement over the performance of the x-ray structure model:
Frame #4853:
Ligand Preparation
Ligand protonation states were assigned using the OpenEye filter program with the -pkanorm option. Compounds with more than 8 rotatable bonds were excluded, and the remaining compounds were prepared using the OpenEye Omega program to generate conformations within an energy window of 5.0 kcal/mol.
Docking
The OEDocking FRED program was used to dock the prepared library into each of the 3 frames identified in the validation study described above. Poses were ranked by the Chemgauss4 score, and visually inspected to eliminate any clearly ugly poses. The top 50-80 compounds in each set should correspond to the steep region of the ROC plots, and ideally should perform with a similar enrichment factor of ~5X.
Links to Fragalysis
https://fragalysis.diamond.ac.uk/viewer/compound_set/DemetriMoustakas-OEDocking/FRED/V04853
https://fragalysis.diamond.ac.uk/viewer/protein_set/DemetriMoustakas-OEDocking/FRED/V04853